Анемия фанкони у детей клинический случай
симптомы, лечение и прогноз жизни
Анемия Фанкони является одной из разновидностей врожденных анемий. На данный момент заболевание полностью не изучено, однако собранные данные позволяют говорить о причинах, проявлении и лечении этой болезни.
История заболевания
Впервые это заболевание было описано в 1937 году швейцарским врачом-педиатром Гвидо Фанкони, который выявил его у трех детей в одной семье. Действительно, заболевание имеет очень много «семейных» случаев. Заболевание это достаточно редкое, в настоящее время в литературе описано более ста случаев заболевания.
Клиника
Анемия Фанкони (врожденная панмиелопатия) – наследственное заболевание, приобретается благодаря аутосомно-рецессивному наследованию, проявляется у гомозигот. Эта болезнь поражает преимущество детей мужского пола. Сопутствующими заболеваниями, причиной которых является анемия Фанкони, может быть предрасположенность к злокачественным новообразованиям (аденома, гепатома, рак пищевода), пороки развития, повышенная чувствительность к химическим мутагенам. У детей, больных анемией Фанкони, существуют дефекты в репарации ДНК, поэтому их хромосомы легко повреждаются даже при малодозном облучении ультрафиолетом.
Заболевание во многом связана с дефектом стволовых клеток, поскольку сыворотка больных, а также их лимфоциты, не оказывают влияния на эффективность колониеобразования во время культивирования донорского костного мозга.
Симптомы
Симптомы анемии Фанкони очень часто не проявляются у малышей до пяти лет, однако при тяжелых случаях болезнь можно диагностировать уже на первом году жизни ребенка. Внешне малыш вялый, на окружающий мир реагирует довольно слабо, у него слабый ответ на внешние раздражители. Кожа больных анемией детей бледная, слизистые оболочки тоже значительно светлее. Заболевание проявляется постепенно, первыми симптомами могут быть кровоподтеки или кровотечения. Помимо этого, больные дети очень подвержены инфекционным заболеваниям по причине ослабленного иммунитета. Селезенка и печень обычно не гипертрофированы, однако лимфоузлы могут увеличиваться, что обычно говорит о наличии инфекции в организме. Лабораторное исследование крови показывает низкое содержание гемоглобина, снижение числа эритроцитов и ретикулоцитов, а вот показатели по лейкоцитам и тромбоцитам в норме. На основании клиники и данных лабораторных исследований врачи могут говорить о наличии у ребенка этого опасного заболевания.
Кроме описанных выше патологических изменений есть еще и другие аномалии, сопровождающиеся при этой болезни. Чаще всего это избыточная пигментация кожи, такие ребятишки отстают в росте. Довольно частым признаком является микроцефалия. Болезнь может вызывать различные патологии развития опорно-двигательного аппарата. Скелет детей, больных анемией Фанкони, может содержать недоразвитые кости – лучевую кость, шейное ребро, кости стоп (велика возможность появления косолапости). Из-за неправильного развития тазовых костей такие младенцы могут родиться с врожденным вывихом бедра.
Если болезнь спровоцировала неврологические проблемы, то среди них чаще всего можно встретить птоз век (опущение), нистагм (дрожание глаз), косоглазие, умственную отсталость, недоразвитость глазных яблок.
В том случае, если анемия Фанкони спровоцировала отклонения в половом развитии, то чаще всего это проявляется в гипоспадии, недоразвитости половых органов, отсутствии одного или двух яичек.
Тяжелые последствие болезни могут выявиться в недоразвитии почек, изменении формы органа (пока принимает форму подковы), наличие в почках множества кист.
Кроме этого, болезнь может спровоцировать развитие пороков сердца.
Лечение
При анемии Фанкони показано стационарное лечение в гематологическом отделении. Стратегия лечения во многом определяется тяжестью течения болезни.
При невозможности помочь больному ребенку консервативными методами показано оперативное вмешательство (спленэктомия), после чего рекомендуется курс анаболических стероидов. Если эти меры не помогают, то необходимо использовать антилимфоцитарный глобулин.
В настоящее время применяется лечение иммунодепрессантами – метотрексатом и циклофосфаном. Однако применение этих препаратов опасно тем, что могут возникнуть кровотечения и появиться воспалительные очаги. Чаще всего эти побочные явления возникают при применении метотрексата, а циклофосфан более щадящий.
Если болезнь перешла в тяжелую стадию, то здесь необходима пересадка костного мозга. Лучше всего, если будет донором однояйцовый близнец. Тогда пересадку можно проводить без иммунодепрессии. Если таких доноров нет, то костный мозг можно брать у сестер или братьев больного ребенка. Перед пересадкой ребенку вводят большую дозу циклофосфана, чтобы не было отторжения.
Трансплантация может сопровождаться тяжелыми осложнениями, когда пересаженный орган (в данном случае костный мозг) будут бороться против реципиента. Это может проявиться в поражении печени и желудочно-кишечного тракта. Донора для пересадки при анемии Фанкони подобрать трудно, поскольку в последнее время семьи не имеют двух и более детей.
Прогноз
При позднем обнаружении анемии Фанкони длительность жизни пациентов не превышает двух-пяти лет. Чаще всего дети умирают от кровоизлияния в мозг или в желудок. Если больному ребенку будет сделана трансплантация костного мозга, то с более, чем пятидесяти процентной вероятностью состояние здоровья ребенка улучшится. Удаление селезенки при незапущенной болезни тоже достаточно эффективно, особенно если было впоследствии проведено лечение анаболическими гормонами. У таких деток может полностью восстановиться функция кроветворения. О хороших прогнозах после лечения болезни можно говорить, если результаты лабораторного анализа крови говорят о нормальной концентрации в крови ее компонентов.
Меня зовут Юлия. Свою жизнь я решила связать с медициной, а именно с педиатрией. Моя любовь к деткам безгранична. Могу сказать, что мне в жизни повезло. Оцените статью: Поделитесь с друзьями!Анемия Фанкони - причины, симптомы, диагностика и лечение
Анемия Фанкони – это генетическое заболевание, которое передается по аутосомно-рецессивному типу и характеризуется нарушением кроветворения, формированием злокачественных новообразований, пороками развития, ломкостью хромосом. Проявляется частыми кровотечениями, кровоподтеками на коже, вялостью, бледностью, склонностью к инфекциям. Диагностика проводится лабораторными методами, назначаются цитогенетическое, молекулярно-генетическое и клиническое исследования крови, миелограмма. Основные способы лечения – пересадка костного мозга, медикаментозное поддержание кроветворения, переливание крови.
Общие сведения
Синонимичные названия анемии Фанкони – врожденная панмиелопатия Фанкони, наследственная панмиелопатия. Заболевание названо по фамилии швейцарского педиатра Гвидо Фанкони, который в 1927 году описал врожденную апластическую патологию на основе симптомов у трех братьев. Анемия Фанкони является редкой генетической болезнью, наследуется согласно аутосомно-рецессивному принципу. Эпидемиологические показатели низкие – 1 больной ребенок на 350 тысяч новорожденных. Распространенность одинакова среди представителей женского и мужского пола, выше в сообществах с разрешенными близкородственными браками, например, у некоторых южноафриканских народов.
Анемия Фанкони
Причины
Заболевание является наследственным, развивается при передаче дефектного гена от родителей к ребенку. Выявлено 15 генов, мутации которых проявляются анемией Фанкони. Из них 14 расположены в аутосомах и являются рецессивными, 1 тип гена находится в X-хромосоме (сцепленной с полом). Все эти гены отвечают за производство определенного фермента, участвующего в репарации ДНК.
Аутосомно-рецессивное наследование подразумевает, что и отец, и мать должны быть носителями патологической генетической информации. При этом сами они, как правило, здоровы. Вероятность рождения больного ребенка в такой паре составляет 25%. Генетическая панмиелопатия диагностируется у детей и взрослых, получивших от каждого из родителей один и тот же измененный ген. В крайне редких случаях анемия провоцируется передачей дефектной Х-сцепленной хромосомы. Женщины могут быть носительницами мутации, заболевание проявляется только у мальчиков. Риск развития патологии у сына при наличии у матери мутированного гена – 50%.
Патогенез
В норме в клетках организма существуют специальные ферментные системы, которые исправляют разрывы молекул ДНК, поврежденных в процессе биосинтеза или воздействия химических, физических реагентов. При анемии Фанкони обнаруживается генетический дефект в кластере белков, ответственных за репарацию ДНК, что приводит к повышенной ломкости хромосом. В итоге у пациентов развиваются нарушения функций костного мозга – неоплазии и апластическая анемия. Онкологические заболевания чаще всего представлены острым миелоидным лейкозом – злокачественной опухолью миелоидного ростка крови, провоцирующей накопление измененных белых клеток, подавляющих рост эритроцитов, тромбоцитов и нормальных лейкоцитов. При апластической анемии в результате дисплазии костного мозга резко угнетается рост и созревание всех трех видов клеток крови.
Симптомы анемии Фанкони
Более чем у половины пациентов наблюдаются врожденные аномалии развития внутренних органов и скелета. Костные деформации проявляются специфическим внешним видом: больные низкорослые, с уменьшенным размером головы, отсутствием или заметным укорочением большого пальца на руках, недоразвитием лучевой кости, врожденным вывихом бедра и/или наличием шейного ребра, косолапостью, недоразвитым подбородком («птичьим лицом»). Характерна гиперпигментация кожи в виде светлых и коричневатых пятен.
Неврологические расстройства представлены косоглазием, недоразвитием одного или двух глаз, опущением верхнего века, глазным дрожанием, глухотой, умственной отсталостью. Больные зачастую имеют незрелые половые органы, у них отсутствует одно или оба яичка. К распространенным аномалиям строения органов относятся пороки мочевыделительной системы: удвоение мочеточников или лоханки, подковообразные почки, почечные кисты, смещенное наружное отверстие уретры (гипоспадия). Врожденные пороки сердца включают атрезию трехстворчатого клапана, дефект межпредсердной перегородки, митральный стеноз, дефект межжелудочковой перегородки. Пациенты страдают от почечной и сердечной недостаточности.
Ключевые симптомы связаны с постепенным нарастанием нарушений в работе костного мозга. Чаще они дебютируют в детском возрасте (в 5-10 лет). Из-за снижения количества тромбоцитов развивается повышенная кровоточивость: при ранениях кровь долго не сворачивается, легко возникают носовые кровотечения, выделения при менструациях обильны, на теле обнаруживается много «беспричинных» кровоподтеков. Уменьшение числа эритроцитов проявляется анемией с характерной слабостью, быстрой утомляемостью, головокружениями, обмороками, бледностью кожи, учащенным сердцебиением и одышкой. Недостаток лейкоцитов способствует ухудшению сопротивляемости инфекциям. Впоследствии формируется лейкоз, миелодиспластический синдром, онкологические болезни.
Осложнения
Наиболее распространенным осложнением считаются частые инфекционные заболевания. У пациентов развивается ОРВИ, ангина, ринит, бронхит, грипп, тиф, герпес. Рецидивирующий характер болезней и их тяжелое течение приводят к деструкции органов, сопровождаются риском сепсиса. Другим осложнением наследственной анемии являются злокачественные новообразования – лейкемия, эпителиальные опухоли органов шеи и головы, половых органов. Рак у таких больных тяжело поддается лечению из-за повышенной ломкости и сниженной репарации ДНК. Это явление ограничивает применение лучевой терапии, цитотоксических препаратов. Нарушение свертываемости становится причиной больших кровопотерь.
Диагностика
Обследование больных проводят онкологи, гематологи, педиатры, врачи-генетики. Диагностика начинается с анализа анамнестических данных и жалоб. Врач выясняет, имеется ли данное наследственное заболевание у близких родственников, уточняет время появления первых признаков болезни, ранние обращения к врачам. При осмотре оценивает общее состояние пациента, выявляет наличие аномалий развития, гиперпигментированных пятен, кровотечений, кровоподтеков. В большинстве случаев не составляет труда обнаружить типичные деформации костей, недоразвитие глаз. Для подтверждения диагноза и различения анемии Фанкони с приобретенной анапластической анемией проводится ряд лабораторных исследований:
- Клинический анализ крови. Характерны изменения клеточного состава крови. На ранних этапах нарушения кроветворения диагностируется тромбоцитопения и лейкопения, на более поздних – панцитопения (резкое снижение объема эритроцитов, лейкоцитов и тромбоцитов). Возможен умеренный гемолиз без гипербилирубинемии, но с ретикулоцитозом. Значение СОЭ увеличено до 60-80 мм/ч.
- Цитогенетическое исследование клеток. Выполняется проба с диэпоксибутаном, митомицином C, указывающая на частоту и спектр хромосомных аберраций. В пользу генетической анемии рассматриваются показатели ДЭБ-теста более 45%, пограничный уровень – 11-45% (процент клеток с хромосомными разрывами).
- Молекулярно-генетический анализ клеток. Исследуются гены, мутации в которых могут привести к развитию заболевания. В 60-70% случаев мутации обнаруживаются в паре генов FANCA, в 14% – в аллели FANCC, в 10% – в генах FANCG. Частота мутаций в других парах – 0,2-3%.
- Миелограмма. По данным исследования определяется увеличение количества плазматических клеток и макрофагов, фагоцитирующих жиры. Содержание недифференцированных клеток – в пределах нормы. Снижена концентрация клеток миелоцитарного ростка, увеличен показатель лимфоцитов.
Лечение анемии Фанкони
Основная терапия направлена на восстановление процесса кроветворения. Методы лечения подбираются индивидуально, зависят от тяжести заболевания, возраста пациента, наличия и выраженности врожденных аномалий. Дополнительно проводится лечение инфекций и онкопатологий, осуществляются реабилитационные мероприятия. Для устранения анемии используются следующие методы:
- ТКМ. Трансплантация костного мозга является наиболее эффективной в долгосрочной перспективе, но имеет противопоказания, нередко сопровождается развитием осложнений. Оптимальный возраст для проведения операции – до десяти лет. Донорами могут выступать здоровые сестра и братья, подходящие по критериям совместимости. Предварительная интенсивная терапия (кондиционирование) связана с риском токсического воздействия на органы. После трансплантации сохраняется высокая вероятность острого или хронического иммунного конфликта между клетками донора и реципиента.
- Медикаментозная стимуляция кроветворения. При невозможности проведения трансплантации пациентам показано консервативное лечение, временно улучшающее их состояние. Выработка кровяных клеток стимулируется андрогенами (мужскими половыми гормонами) и гематопоэтическими факторами роста – эритропоэтином, фактором стволовых клеток, интерлейкинами-1-12. Параллельно применяются иммунодепрессанты. Медикаментозная терапия способна на протяжении многих лет поддерживать высокое качество жизни больных, но ее эффективность постепенно снижается.
- Переливание компонентов крови. При выраженных побочных эффектах или противопоказаниях к этиотропной терапии (трансплантации, стимуляции кроветворения) назначаются процедуры гемотрансфузии. Переливаются отмытые эритроциты – донорские красные кровяные тельца, освобожденные от поверхностных белков. При кровотечениях и снижении уровня тромбоцитов пациентам вводится тромбоцитарная масса.
Прогноз и профилактика
Продолжительность жизни больных определяется степенью нарушения функции костного мозга. Иногда пациенты доживают до 40 лет без лечения, но нередко умирают в детстве от тяжелой анемии или онкологических заболеваний. Прогноз наиболее благоприятен при своевременном проведении аллогенной трансплантации костного мозга, после которой есть шанс полного восстановления нормального кроветворения и увеличения срока жизни. Поскольку заболевание генетическое, предотвратить его развитие невозможно. Профилактика сводится к медико-генетической консультации супружеских пар из групп риска, планирующих беременность, а также к проведению пренатальной диагностики патологии, в ходе которой из пуповинной вены плода производится забор крови и выполняется ДЭБ-тест. При его положительном результате рассматривается вопрос о прерывании беременности.
Клинический случай апластической анемии Фанкони Текст научной статьи по специальности «Клиническая медицина»
DOI: 10.24411/2074-1995-2019-10029
Клинический случай апластической анемии
Фанкони
Т.А.Емельянова1, Т.А.Миненкова1, А.В.Серёжкина1, Н.С.Разинькова1, И.Г.Хмелевская1, Г.М.Сычева2, Н.В.Чаплыгина2, Т.В.Феоктистова2 Журский государственный медицинский
университет
2Областная детская клиническая больница,
Курск
Анемия Фанкони (АФ) - редкое аутосомно-рецес-сивное заболевание, характеризующееся множественными врожденными физическими аномалиями, прогрессирующей костно-мозговой недостаточностью и предрасположенностью к развитию злокачественных новообразований. Клетки всех больных АФ характеризуются гиперчувствительностью к ДНК-повреждающим агентам, что определяет нестабильность генома. В статье описан клинический случай анемии Фанкони у ребенка 6 лет и результат успешной трансплантации гемопоэтических стволовых клеток костного мозга.
Ключевые слова: анемия Фанкони, стволовые клетки, дети, гематология, аллогенная трансплантация.
Clinical Case of Fanconi Aplastic
Anemia
T.A. Emelyanova1, T.A.Minenkova1, A.V.Seryozhkina1, N.S.Razinkova1, I.G.Khmelevskaya, G.M.Sycheva2, N.V.Chaplygina2, T.V.Feoktistova2 :Kursk State Medical University, Kursk 2Regional Children's Clinical Hospital, Kursk
Fanconi anemia (FA) is a rare autosomal recessive disorder, manifested by multiple congenital physical abnormalities, progressive bone marrow failure, and a predisposition to the development of malignant neoplasms. The cells of all patients with FA have a certain hypersensitivity to DNA-damaging agents, which determines the instability of the genome. The article describes a clinical case of a 6-year-old child with FA and the result of successful transplantation of hematopoietic stem cells of the bone marrow.
Keywords: Fanconi anemia, stem cells, children, hematology, allogeneic transplantation.
Анемия Фанкони является редким врожденным заболеванием с аутосомно-рецессивным типом наследования, вариабельной пенетрантностью и генетической гетерогенностью [1]. Заболевание характеризуется нестабильностью геномного аппарата, врожденными аномалиями развития и нарушением гемопоэза, а также повышенной склонностью к развитию опухолевых заболеваний [2].
В настоящий момент в литературе имеется описание более 2 тыс. случаев анемии Фанкони у детей и к настоящему времени их количество быстро увеличивается, благодаря внедрению методов лабораторной диагностики, позволяющих установить диагноз у сиблингов больного анемией Фанкони еще до манифестации апластической анемии, а так же у больных с характерными пороками развития, но без гематологических аномалий [3].
Частота встречаемости данной патологии составляет 1:360 тыс. родившихся детей, с соотношением 1,1:1 в пользу мальчиков [1].
Впервые анемия Фанкони описана в 1927 г. швейцарским педиатром Гвидо Фанкони как семейный случай панцитопении у трех братьев с физическими пороками развития [4]. В 1931 г. Нигели был предложен термин «анемия Фанкони» для обозначения комбинации семейной апластической анемии и врожденных физических пороков [5].
Генетической основой анемии Фанкони являются 19 ассоциированных с заболеванием генов, продуцирующих белки, которые участвуют в патогенезе заболевания. Один из них - FANCB - находится в Х-хромосоме, остальные - на аутосомах [6].
При данной форме анемии выявлено более быстрое укорочение теломераз в клетках крови, что играет определенную роль в генетической нестабильности клеток [7, 8]. Мутировавшие гены продуцируют белки, взаимодействующие с протеинами, которые участвуют в репарации повреждений ДНК [5]. Нарушение процесса репарации клеток является одной из важных причин повышенной чувствительности клеток к ДНК-повреждающим воздействиям и обуславливает наклонность к развитию опухолевых процессов у данной категории больных [9].
При анемии Фанкони клетка теряет способность исправлять определенный тип повреждений ДНК-поперечных межхроматидных сшивок, препятствующих работе репликационной вилки [10].
Данное заболевание имеет полиморфную клиническую картину, средний возраст установления диагноза анемии Фанкони составляет 7,5 лет у мальчиков и 9 лет у девочек, в 75% случаев диагностируется в возрасте 4-12 лет, когда появляется гематологическая симптоматика [3].
Наряду с проявлениями аплазии кроветворения и цитопении отмечаются физические аномалии. Уже с рождения клинически характерна задержка физического развития, сохраняющаяся в дальнейшем. У части больных костный возраст отстает на 5-7 лет от паспортного.
К основным симптомам анемии Фанкони относится задержка физического развития, имеющая пропорциональный характер: отставание в росте, массе, микроцефалия, микрофтальмия, гипогона-дизм, крипторхизм [9]. Специфичный признак -аномалии развития верхних конечностей: аплазия или гипоплазия большого пальца кисти, синдактилия, косорукость, уменьшение количества костей запястья, отсутствие и недоразвитие лучевой кости и др. [7]. Встречаются аномалии нижних конечностей и позвоночника (косолапость, врожденный вывих бедра, сколиоз, кифоз и др. [10]. Могут быть аномалии глаз (стробизм, эпикантус, птоз века, гир-пертелоризм).
Характерны пороки мочевыделительной системы (аплазия почки, подковообразная почка, мегауретер и др.), половой системы, врожденные пороки сердца, пищеварительной системы, органов дыхания. У некоторых больных имеются изменения ЦНС, представленные косоглазием, глухотой, умственной от-
н
е
ц а
ы н
нду р
о
нн
1—
<
1—
<
О.
to
z
<
сс
1—
и
нн
iii
ш 15
о
—X
—X
<
ос
s
<
1—
<
и
<
о_
1—
ОС
<
ш
LU i_
о
<
о
о
I—
го
-О
.CP
сталостью или замкнутостью, психической инфантильностью, реже - дебильностью [6].
Около 10-33% больных не имеют врожденных пороков развития [8].
У большинства больных отмечена пигментация на коже и слизистых оболочках диффузного или очагового типа с участками слияния в естественных складках и в местах, защищенных от солнца, различной степени выраженности и цвета от «кофе с молоком» до почти черных участков. Изменения на коже появляются у некоторых больных с рождения, у других предшествуют развитию гематологической симптоматики [7].
Заболевание имеет хронический характер, протекает с чередованием периодов ремиссии и обострения, медленно прогрессирует.
По мере взросления у людей с анемией Фанкони прогрессирует трехростковая апластическая анемия, подавляются все функции костного мозга [2]. Чаще в дебюте гематологических проявлений лидирует тромбоцитопения, проявляющаяся спонтанными эк-химозами и петехиальной сыпью, носовыми кровотечениями с последующим присоединением прогрессирующей анемии и лейкопении. Выраженная панцитопения отсутствует у детей 3-8 лет. Анемия имеет нормохромный характер, сопровождается анизоцитозом, макроцитозом, умеренным пойкило-цитозом [4]. Типичным при апластической анемии является повышение фетального гемоглобина вкупе с макроцитозом [9]. Лейкопения обычно стойкая, достигает максимального уровня в терминальном периоде. В костномозговом пунктате отмечается снижение клеточности с относительным преобладанием эритроцитопоэза, лимфоцитов, встречаются плазматические, тучные клетки и стромальные элементы. В биоптатах костного мозга на ранних стадиях заболевания встречаются гиперклеточные участки активного резидуального гемопоэза, которые исчезают по мере прогрессирования заболевания. В последующем развивается гипоплазия костного мозга [6].
Средняя продолжительность жизни детей с момента появления гематологической симптоматики составляет в среднем 6 лет. Причиной летального исхода являются тяжелые кровотечения и кровоизлияния во внутренние органы и развитие инфекционных осложнений [4].
Около 10% пациентов, описанных в литературе, в дебюте заболевания развернули картину острого лейкоза и миелодисплатического синдрома [7]. Согласно исследованиям международного регистра анемии Фанкони, риск развития острого миелобо-ластного лейкоза и миелодиспластического синдрома у больных с анемией Фанкони равен 52% к 40 годам.
Кроме гематологических аномалий, для больных анемией Фанкони характерна склонность к развитию опухолей. Риск развития злокачественных опухолей у этой категории больных составляет 10%, из них 5% приходится на долю опухолей печени и 5% -на остальные опухоли [6]. У детей опухоли встречаются редко, средний возраст диагностики составляет 16 лет.
«Золотым стандартом» скрининга для выявления анемии Фанкони является тест диэпоксибутаном (ДЭБ, DEB, 1,3-butadienediepoxide) и его вариант с митомицином С (ММС) [4]. Одним из феноменов, характерных для клеток больных анемией Фанкони является их склонность к формированию специфических хромосомных аномалий (разрывов, сестринских обменов, эндорепликаций) при культивировании клеток in vitro [1]. Инкубация стимулированных
лимфоцитов больных с алкилирующими агентами резко увеличивает количество аберраций. Данный кластерный эффект лежит в основе диагностики и дифференциальной диагностики анемии Фанкони. Под влиянием бифункциональных алкилирующих агентов происходит замедление клеточного цикла -клетки больных анемией Фанкони останавливаются в G2 фазе митотического цикла, в связи с этим, разработан еще один тест диагностики - проточная ци-тофлоуметрия [8].
На современном этапе существуют две возможности коррекции апластической анемии Фанкони: замещение недостающего пула стволовых клеток донорскими и снятие ингибиции пролиферации рези-дуальных стволовых клеток [10].
На настоящий момент аллогенная трансплантация костного мозга является единственным методом окончательного лечения гематологического синдрома при анемии Фанкони. Оптимальный возраст для проведения операции - до десяти лет. Донорами могут выступать здоровые сестры и братья, подходящие по критериям совместимости и «молекулярно» совместимые неродственные доноры. Эффективность проведения трансплантации составляет 75-90% полного выздоровления [5].
Симптоматическое лечение включает андрогены -препараты, способствующие улучшить краткосрочный и долгосрочный прогноз при анемии Фанкони, но не способные коренным образом изменить прогноз заболевания [9]. На лечении андрогенами гематологический ответ достигается у 50% больных [7]. После наступления резистентности к андрогенам развивается прогрессия цитопении, осложнения которой наряду с лейкемией и солидными опухолями являются главной причиной смерти пациентов [9].
Необходимые методы лечения подбираются индивидуально, определяются тяжестью течения заболевания, возрастом пациента, наличием аномалий развития и их выраженности [4].
В качестве примера мы приводим клинический случай апласической анемии Фанкони, диагностированной у ребенка 6 лет.
В гематологический стационар областной детской больницы г. Курска (ОДКБ) поступил ребенок Николай Ю. с жалобами на изменения в анализах крови в виде анемии, снижения уровня тромбоцитов и носовые кровотечения.
Из анамнеза жизни известно, что ребенок рожден от III беременности, протекавшей на фоне хронической фетоплацентарной недостаточности и внутриутробной гипоксии плода, задержки внутриутробного развития плода II степени, III преждевременных оперативных родов на сроке 35 нед. по поводу несостоятельности рубца на матке. Масса при рождении -1900 г., длина тела - 44 см. Оценка по шкале Апгар -7 баллов. В возрасте 1 мес. переведен в отделение патологии новорожденных с диагнозом: врожденный порок сердца: открытый артериальный проток, открытое овальное окно, стеноз легочной артерии, НК 1 ст. гипотрофия 1-2-й степени. Пре- и перинатальное поражение ЦНС. Синдром тонусных нарушений. Ретинопатия недоношенных 0-1-й степени. Полидактилия обеих кистей. Внутриутробная инфекция. Консультирован в НЦССХ им. А.Н.Бакулева, оперативная коррекция порока не проводилась. Данные о дальнейшем динамическом развитии ребенка отсутствуют, в связи с неоднократной переменой родителями места жительства и утратой медицинской документации.
23.03.2018 амбулаторно у ребенка отмечалось обильное носовое кровотечение, с 27.03 по 05.04.2018 г. на-
Рис. 1. Стигмы дисэмбриогенеза при анемии Фанкони -клювовидная деформация носа, низко посаженные уши.
ходился на лечении в Областной инфекционной больнице им. Семашко г. Курска по поводу острой правосторонней интерстициальной пневмонии средней степени тяжести. Во время госпитализации рецидивировали носовые кровотечения, в лабораторном контроле наросли явления анемии (Нв - 88-75 г/л). С подозрением на постгеморрагическую анемию доставлен в гематологический стационар ОДКБ 05.04.2018.
При осмотре: выражена задержка физического (возраст 6 лет: рост - 118 см, масса тела - 20 кг) и нервно-психического развития. Пониженного питания. Обращает на себя внимание характерный фенотип пациента: кожные покровы смуглые с желтушным оттенком (пациент цыган), пигментация скул, периорбитальной и периоральной областей, области шеи, боковых поверхностей туловища со сгущением темного оттенка до черного в области естественных складок, на коже туловища единичные пятна цвета «кофе с молоком», геморрагических элементов на коже и слизистых оболочках не отмечено. Черепно-лицевые признаки: зауженное небо, клювовидная деформация носа, низко посаженные уши, птоз обеих век, монголоидный разрез глаз (рис. 1).
Имеет место удвоение ногтевой фаланги обеих первых пальцев рук (рис. 2). Периферические лимфатические узлы не увеличены. Над всей областью сердца грубый систолический шум, ЧСС - 100 в минуту, АД - 100/60 мм рт. ст. Живот мягкий, безболезненный, печень и селезенка не увеличены. Отмечается ночной энурез.
Проведено динамическое лабораторное обследование, в результате которого выявлено нарастание анемического синдрома в виде нормохромной анемии от легкой до тяжелой степени (Нв - 63 г/л, эритроциты 1,83х1012/л), требующей проведения гемо-трансфузии, тромбоцитопении до единичных клеток в препарате, нуждающейся в заместительной терапии.
В миелограмме: костный мозг гипоклеточный, тип кроветворения нормобластический, бласты 0,4 (левая точка) и 1,8% (правая точка), угнетение всех ростков кроветворения: гранулоцитарный: слева 4,2%, справа 9%, эритроидный: 7% слева и 18,4% справа, в препа-
ратах присутствуют по 1 мегакариоциту, без отшну-ровки тромбоцитов. В препарате жировые пустоты, скопления из ретикулярых и стромальных клеток эритроидного ряда и единичных нейтрофилов. Присутствуют митозы клеток эритроидного ряда.
Рентгенограмма костей кистей рук: удвоение ногтевых фаланг первых пальцев обеих кистей, развитие костей кистей соответствует 2 годам.
УЗИ сердца: пролапс правой створки митрального клапана без нарушения функции клапана и гемоди-намических нарушений.
УЗИ почек: ротационная дистопия правой почки.
Консультация невропатолога: резидуальное поражение ЦНС. Легкие когнитивные нарушения. Ночной энурез.
Консультация генетика: синдром Рубинштейна-Тейби.
24.04.2018 амбулаторно проведена консультация гематолога ФГБУ НМИНЦ ДГОИ им. Д. Рогачева г. Москва, с проведением ДЭБ-теста в лаборатории ци-тогенетики и молекулярной генетики: исследовано 30 метафаз, выявлено 108 аббераций. ДЭБ-тест положительный.
На основании проведенного обследования поставлен диагноз: D 61.3 Врожденная апластическая анемия Фанкони. Синдром Рубинштейна-Тейби. Пролапс митрального клапана без нарушения гемодинамики. Резидуальное поражение ЦНС, легкие конгитивные нарушения. Ночной энурез. Отставание в физическом развитии, дефицит массы тела. Ротационная дистопия правой почки.
Проведено типирование на HLA-совместимость возможных доноров (сиблингов: родного брата и сестры), результат исследовании отрицательный.
В настоящий момент отмечается трансфузионная зависимость от компонентов крови, госпитализации ежемесячно, в связи с ухудшением состояния.
Кровь ребенка находится в банке для подбора донора для трансплантации костного мозга (ТКМ). 29.10.10 мальчик госпитализирован в стационар кратковременного лечения ФГБУ НМИНЦ ДГОИ им. Д.Рогачева для проведения аллогенной трансплантации гемопоэтических стволовых (ТГСК). Проведено предтрансплантационное обследование перед трансплантацией гемопоэтических стволовых клеток от неродственного полностью совместимого донора с ТСИаЬ деплецией (ПСКК). Абсолютных противопоказаний к ТГСК не выявлено. С 08.11.18 по 13.11.18 проведено кондиционирование по схеме ТАО 2 Гр (08.11.18) + Тимоглобулин 5 мг/кг + Эндок-сан 40 мг/кг + Мабтера + Флударабин 150 мг/м2. Кондиционирование перенес удовлетворительно на фоне сопроводительной терапии. Миелоинфузия от 14.11.18: Ж!/кг (х108): 2,80, CD34+ (х106/кг): 13,07, ТСИаЬ+ (х103/кг): 0,28, CD3+(х106/кг): 5,80, CD19+ (х106/кг): 0,05, CD56+ (х106/кг): 23,27. На девятые сутки у ребенка зафиксировано приживление лейкоцитарного ростка (лейкоциты 1,27 тыс./мкл). Проведена профилактика РТПХ - циклоспорин с первых суток. Отмечалось проявление осложнений после ТГСК: мукозит потребовавший обезболивания тра-мадолом, почечная токсичность на фоне терапии циклоспорином (нормализация показателей после паузы в 5 дней, доза уменьшена по 50%). В дальнейшем динамика положительная, состояние ребенка за время пребывания в СКЛ стабильное. Инфекционных осложнений не было. Проявлений РТПХ не было. Концентрация циклоспорина удовлетворительная, признаков нефротоксичности нет. В стабильном состоянии, на сроке 60 сут. после ТГСК ребенок выписан для продолжения терапии по месту жительства.
о
го
.а
.сх
В последние годы анемия Фанкони является предметом интенсивных исследований, прежде всего в области репарации ДНК. Многие открытия привели к пониманию канонического пути, где все протеины, связанные с развитием АФ, функционируют последовательно в различных комплексах для репарации повреждений межнитевых поперечных сшивок ДНК. Развиваются и исследования детальной архитектуры этих путей. Вопрос о том, как дефектный процесс репарации ДНК приводит к тому или иному фенотипу, пока до конца не решен. Для оптимизации диагностики АФ все пациенты должны быть направлены на генетическое консультирование совместно с их родителями и родственниками. Анализ на наличие мутаций должен выполняться всем сиб-лингам пациента, особенно при планировании трансплантации стволовых кроветворных клеток, а также в семьях, планирующих иметь детей.
Анемия Фанкони имеет относительно неблагоприятный прогноз. Продолжительность жизни больных определяется степенью нарушения функции костного мозга. Прогноз наиболее благоприятен при своевременном проведении аллогенной трансплантации костного мозга, после которой есть шанс полного восстановления нормального кроветворения и увеличения срока жизни [5].
Литература
1. Панферова А.В., Тимофеева, Н. М., Ольшанская, Ю.В.. Генетическая диагностика анемии Фанкони. Обзор литературы. Он-когематология. - 2010. - Т. 11. - С. 76-83. / Panferova A.V., Tim-ofeeva, N. M., Ol'shanskaya, Yu.V.. Geneticheskaya diagnostika anemii Fankoni. Obzor literatury. Onkogematologiya. 2010; 11: 76-83. [in Russian]
2. Auerbach A.D. Fanconi Anemia and its Diagnosis. Mutat Res. 2009; 668: 1-2.
3. Емельянова Т.А., Миненкова Т.А., Яковлева А.В., Хмелевская И.Г., Сычева Г.М., Чаплыгина Н.В., Феоктистова Т.В., Фисюн
И.В. Клинический случай врожденного дискератоза. Трудный пациент. - 2018. -Т. 16. - № 5. - С. 41-43. / Emel'yanova T.A., Mi-nenkova T.A., Yakovleva A.V., Khmelevskaya I.G., Sycheva G.M., Chaplygina N.V., Feoktistova T.V., Fisyun I.V. Klinicheskiy sluchay vrozhdennogo diskeratoza. Trudnyy patsient 2018; 16: 5: 41-43. [in Russian].
4. Fancony Anemia: Guidelines for diagnostic and manegment. Fourth edition 2014 Fanconi Anemia Research Fund, Inc., Eugene, Oregon, 2014.
5. Дикин Д.С., Мазин А.В. Анемия Фанкони: на перекрестке репарации ДНК (обзор). Биохимия. - 2011. - Т. 76. - № 1. - С. 4661. / Dikin D.S., Mazin A.V. Anemiya Fankoni: na perekrestke reparatsii DNK (obzor). Biokhimiya. - 2011. -76. - № 1. - 46-61. [in Russian].
6. Kitao H., Takata M. Fanconi anemia: a disorder defective in the DNA damage response. Int. J. Hematol. 2011; 93 (4): 417-424.
7. Самочатова Е.В., Масчан А.А., Кравченко Е.Г., Новичкова Г.А., Ско-робогатова Е.В., Масчан М.А., Стобецкий В.И., Румянцев А.Г. Анемия Фанкони: диагностика и терапия. Вопросы гематологии/онкологии и иммунопатологии в педиатрии. - 2002. - Т. 1. - № 1. -С. 12-17. / Samochatova E.V., Maschan A.A., Kravchenko E.G., Novichkova G.A., Skorobogatova E.V., Maschan M.A., Stobetskiy V.l., Rumyantsev A.G. Anemiya Fankoni: diagnostika i terapiya. Voprosy gematologii/onkologii i immunopatologii v pediatrii. 2002; 1: 1: 12-17. [in Russian].
8. Румянцев А.Г. Трансплантация гемопоэтических стволовых клеток у детей. А.Г.Румянцев, А.А.Масчан. - М.: МИА, 2003. / Rumyantsev A.G. Transplantatsiya gemopoeticheskikh stvolovykh kletok u detey. A.G.Rumyantsev, A.A.Maschan. M.: MIA, 2003. Rumyantsev A.G. Transplantatsiya gemopoeticheskikh stvolovykh kletok u detey. A.G.Rumyantsev, A.A.Maschan. - M.: MIA, 2003 [in Russian].
9. Vanuytsel K., Cai Q., Nair N. et al. FANCA knockout in human embryonic stem cells causes a severe growth disadvantage. Stem Cell Res. 2014; 13 (2): 240-250. [in Russian].
10. Rogers K. J., Fu W., Akey J. M., Monnat R. J. Global and disease-associated genetic variation in the human Fanconi anemia gene family. Hum Mol Genet. 2014; 23 (25): 6815-6825.
О
О I—
NÍ
X
J
ro
Сведения об авторах:
Емельянова Татьяна Александровна - к.м.н., ассистент кафедры педиатрии ГБОУ ВО КГМУ Минздрава России, Курск Миненкова Татьяна Александровна - к.м.н., ассистент кафедры педиатрии ГБОУ ВО КГМУ Минздрава России Курск Серёжкина Александра Владимировна - ассистент кафедры педиатрии ГБОУ ВО КГМУ Минздрава России, Курск Разинькова Наталья Сергеевна - к.м.н., доцент кафедры педиатрии ГБОУ ВО КГМУ Минздрава России, Курск Хмелевская Ирина Григорьевна - д.м.н., профессор, зав. кафедрой педиатрии ГБОУ ВО КГМУ Минздрава России, Курск
Сычева Галина Михайловна - главный внештатный детский нефролог комитета здравоохранения Курской области, заведующая отделением гематологии и нефрологии ОБУЗ ОДКБ, Курск
Чаплыгина Наталья Васильевна - главный внештатный детский гематолог комитета здравоохранения Курской области, врач -педиатр отделения гематологии и нефрологии ОБУЗ ОДКБ
Феоктистова Татьяна Васильевна - врач -педиатр отделения гематологии и нефрологии ОБУЗ ОДКБ, Курск
Анемия Фанкони у детей: симптомы, причины и лечение
Анемия Фанкони – это крайне редкое заболевание, являющееся следствием врожденного генетического отклонения. Встречается болезнь преимущественно у жителей Южной Африки и евреев-ашкенази не чаще, чем 1 случай на 350 тысяч новорожденных детей. Впервые патология была описана в 1927 году педиатром Гвидо Фанкони. По последним данным, зарегистрировано 1200 случаев этого заболевания.
Механизм развития
Причины анемии Фанкони кроются в генетических отклонениях. Патология развивается по аутосомно-рецессивному механизму наследования. Данное понятие подразумевает наследование поврежденного гена ребенком одновременно от матери и отца. Если измененный ген был только у одного из родителей, болезнь не развивается, но такой человек становится носителем мутации и сможет передавать измененный ген своим детям.
Реже случается болезнь с рецессивным Х-сцепленным механизмом наследования. Здесь анемия передается сыну от матери с вероятностью 50%. Кроме основных симптомов патологии, у детей часто возникают и другие генетические отклонения.
Симптомы
Симптомы анемии Фанкони длительное время отсутствуют, первые проявления наблюдаются у пациентов, достигших шести лет. На врожденное заболевание указывают такие внешние признаки:
- низкий рост, ребенок значительно отстает от своих сверстников. Особо заметным становится этот признак при поступлении в школу;
- микрофтальмия – отклонение, характеризующееся меньшим размером одного из глазных яблок. Чаще встречается односторонний тип, но есть случаи и двусторонней микрофтальмии;
- микроцефалия – недоразвитость черепной коробки, сопровождающаяся умственным отставанием. При этом неправильно формируются черепные кости, наблюдается раннее схождение родничка;
- темный оттенок кожи;
- дефект формирования больших пальцев на руках, искажение суставов и утрата работоспособности;
- пигментные пятна на теле.
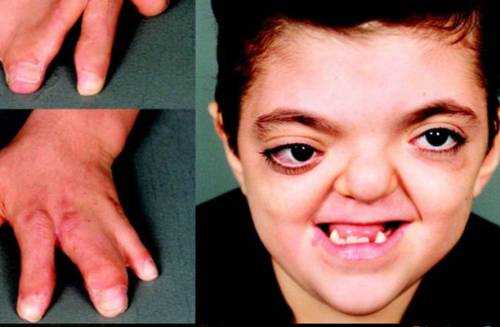
Болезнь вызывает тяжелые внешние и внутренние дефекты
Дети с анемией Фанкони сильно отстают в развитии, часто у них диагностируется глухота, дрожание глаз, одно веко опущено, аномальное строение полового члена у мальчиков, отсутствие яичек. Нередко у таких пациентов обнаруживаются различные сердечно-сосудистые пороки. Во время обследования выявляется гипоплазия или поликистоз почек, мочеточники и лоханки удвоены, орган имеет подковообразный тип.
Прогрессирование заболевания постепенно нарушает функцию костного мозга, что негативно сказывается на процессе кроветворения. У пациентов 2-х – 3-х лет значительных отклонений в составе крови не наблюдается, кроме макроцитоза, – увеличения размера эритроцитов. После 5–7 лет количественный и качественный состав крови значительно ухудшается. Это вызывает такие симптомы:
- бледность кожных покровов из-за снижения количества эритроцитов;
- быстрая утомляемость ребенка, снижение успеваемости, мышечная слабость;
- приступы тахикардии, отдышки, нехватки воздуха.
Больные с анемией Фанкони подвержены частым вирусным и бактериальным болезням, которые протекают с осложнениями.
Важно! На фоне патологии у многих детей развиваются онкологические болезни, включая лейкоз, миелодиспластический синдром.
Диагностика
Рассматриваемое заболевание легко распознать внешне, но несмотря на это, для постановки диагноза необходимо проведение лабораторной диагностики. Для больных характерен такой феномен, как склонность кровяных клеток к образованию специфических хромосомных отклонений. In vitro обнаруживаются разрывы, сестринские обмены, эндоредупликации и прочие нарушения. Такой феномен на уровне ДНК получил название в медицинской практике – кластогенный эффект. Именно это помогает провести диагностику и дифференциальную диагностику анемии.
Еще один современный метод выявления патологии – флюорометрия. Известно, что у пациентов с рассматриваемым заболеванием клеточный митоз останавливается посредине фазы развития клеток. С помощью этого метода удается быстро и достоверно установить диагноз, начать необходимую терапию.

Диагностика заболевания проводится с помощью лабораторного исследования анализа крови и некоторых других методов
Для больных характерны такие нарушения в составе крови:
- лейкопения – снижение количества лейкоцитов;
- тромбоцитопения – уменьшение уровня тромбоцитов;
- панцитопения – общее нарушение состава крови со снижением концентрации всех элементов;
- снижение ретикулоцитов;
- макроцитоз – увеличение размера эритроцитов;
- рост фатального гемоглобина.
Особое внимание уделяется исследованию костного мозга. В первую очередь диагностика проводится среди детей, перенесших раковые заболевания крови. Часто после успешного излечения лейкоза у ребенка через несколько лет обнаруживалась анемия Фанкони.
Анализ пункции костного мозга показывает преобладание лимфоцитов, обеднение кроветворных элементов, наличие плазматических, стромальных и тучных клеток. Но эти признаки присущи также и апластическому виду малокровия, поэтому окончательный диагноз ставят лишь после комплексного генетического тестирования.
Последствия
Анемия этого типа считается тяжелым неизлечимым заболеванием, ведущим к развитию многих последствий. В большинстве случаев даже при грамотном лечении продолжительность жизни пациента не превышает 30 лет. К распространенным последствиям следует отнести:
- тяжелое течение грибковых, вирусных, бактериальных болезней с развитием осложнений, поражением всех внутренних органов и систем. Ребенок часто болеет, даже обычная простуда вызывает отиты, бронхиты и прочие последствия;
- из-за дефицита тромбоцитов возникает кровоточивость десен, частые кровотечения из носа;
- формирование злокачественной опухоли крови с быстрым прогрессированием патологии. Справиться с онкологией удается в крайне редких случаях.
Значительно повышен у людей с анемией Фанкони риск развития онкологии. Согласно статистке, эта угроза выше на 10 % чем у обычных людей. Половина случаев характеризуется поражением печени. Сюда относят гепатоцеллюлярную карциному, гепатому, аденому и другие. Чаще рак обнаруживается у больных после достижения 16 лет, преимущественно у мальчиков. У девочек диагностируются онкологические образования других внутренних органов (рак языка, пищевода).
Методы лечения
Консервативное лечение заболевания носит лишь симптоматический характер. Ни один из методов терапии не способен в корне изменить исход патологии. Для облегчения состояния пациента в медицине используют такие медикаментозные средства, как андрогены. В конце 50-х годов это лекарство было впервые использовано среди пациентов с анемией Фанкони.
Медикаментозная терапия направлена на устранение симптомов, замедление хода развития патологии
В этой сфере популярностью пользуется средство под названием Оксиметалон. Этот анаболический стероид способствует увеличению мышечной массы человека, катализирует синтез костным мозгом красных кровяных телец, повышает эффективность других стероидов. Эффект от использования препарата отмечается через 1,5–2 месяца. В этот период у больного увеличивается концентрация в крови лейкоцитов, тромбоцитов, эритроцитов. Численность тромбоцитов растет очень медленно. Обычно для достижения желаемого лечебного результата требуется не менее года. После прекращения приема медикамента у большинства больных регистрируются рецидивы анемии. В крайне редких случаях повторного обострения не происходит, что связано с наступлением полового созревания.
Использование андрогенов способствует увеличению продолжительности жизни больного. Обычно пациенты, принимающие такие средства, живут на 9 лет дольше. Ранее в комплексе с андрогенами использовали глюкокортикостероиды. Широко применялось средство Преднизолон. Но на сегодняшний день доказано, что этот препарат в роли самостоятельной терапии не имеет лечебной ценности.
Большую сложность составляет проблема терапии лейкемий и миелодиспластических синдромов у людей с данной патологией. У таких пациентов чувствительность клеток к химиотерапии снижена, а из-за повышенной ломкости хромосом развиваются тяжелые последствия. Подавляющее количество больных с онкологическими болезнями даже после проведения соответствующего лечения погибает. Летальный исход наблюдается спустя 2–3 месяца после постановки диагноза. Крайне редко химиотерапия помогает справиться с раком. При этом средняя продолжительность жизни таких людей соответствует продолжительности жизни других больных с анемией Фанкони.
Важно! Медикаментозное лечение является лишь поддерживающим, для людей, проходящих такую терапию, прогноз чаще неблагоприятный.
Хирургическое вмешательство
Единственным способом полного избавления ребенка от генетического заболевания считается оперативное вмешательство. С этой целью проводится аллогенная трансплантация гемопоэтических стволовых клеток костного мозга.
Выполнение операции осуществляется при наличии подходящего донора. При этом всегда существует риск развития негативных последствий, например, отторжение новых клеток, ведь они воспринимаются организмов в роли чужеродных тел.
Хирургическое вмешательство не всегда дает ожидаемый терапевтический эффект
В связи с дороговизной и недостаточностью квалифицированных кадров в южноафриканских континентах таких операций проведено не более 250 среди всех больных.
Другие направления лечения
Кроме использования андрогенов, лечебная тактика включает и другие направления, среди которых можно выделить:
- назначение гормональных средств, которые выступают в роли катализатора нормального функционирования костного мозга;
- оперативное вмешательство по удалению селезенки;
- применение глобулина антилимфоцитарного типа;
- переливание крови;
- назначение иммунодепрессантов (Метотрексат, Циклофосфан).
В большинстве случаев эти способы помогают лишь продлить жизнь пациента. Полное выздоровление возможно лишь при пересадке костного мозга.
Прогноз
Анемия Фанкони – это редкая генетическая патология, которая не поддается консервативному лечению. Чаще для пациентов прогноз неблагоприятный. Даже трансплантация костного мозга помогает лишь в 50% случаев. При развитии у человека лейкоза и других сопутствующих заболеваний прогноз значительно ухудшается.
На фоне сильного угнетения иммунитета, развития хронических кровотечений, онкологических болезней и других осложнений в большинстве случаев наступает смерть больного. Средняя продолжительность жизни составляет около 30 лет.
что это, симптомы и лечение
Одним из самых серьезных генетических заболеваний, проявляющихся в детском возрасте, является анемия Фанкони. Первые упоминания об этой болезни появились в 1927 году. Ее открыл педиатр из Швейцарии, по имени которого она и названа. Чтобы понять какой прогноз дается при этом заболевании нужно разобраться в его особенностях и механизме развития.
Анемия Фанкони – что это?
Анемия Фанкони или, как ее еще называют, врожденная панмиелопатия является наследственной патологией, появляющейся в результате рецессивного наследования. Этому заболеванию свойственны нарушения в работе кроветворной системы. Анемия Фанкони провоцирует образование злокачественных опухолей, пороки в развитии. Хромосомы у детей, страдающих данной патологией, становятся ломкими. Недуг проявляет себя частыми кровотечениями, делает больного восприимчивого к разным инфекциям.

Встречается эта патология нечасто, ей в большей степени подвержены мальчики.
Причины анемии Фанкони у детей
Болезнь проявляется у детей под влиянием генетической мутации, наследуемой по аутосомно-рецессивному пути. При наличии идентичного дефектного гена у матери и отца, вероятность появления младенца с Анемией Фанкони равна 25%.
На сегодняшний день обнаружено 15 генов, изменения в которых провоцируют панцитопении. Один локализуется на Х-хромосоме, а другие в аутосомах. За счет генов кодируются белки, восстанавливающие ДНК.
Мутации в них чреваты следующими последствиями:
- Изменениями в составе стволовых клеток.
- Чрезмерной ломкостью хромосом.
- Ухудшением процессов кроветворения.
- Увеличением продолжительности вызревания клеток, отвечающих за кроветворение.
- Ускорением отмирания эритроцитов.
Симптомы
В большинстве случаев симптоматика анемии Фанкони начинает проявлять себя при достижении детьми возраста 4-10 лет. Список самых распространенных симптомов включает:
- внезапные кровотечения;
- появление кровоподтеков на кожных покровах;
- побледнение кожи и слизистых;
- ослабление реакций на внешние раздражители.
Еще один характерный признак – это восприимчивость к инфекциям. В некоторых случаях имеет место лимфаденопатия. А вот печень с селезенкой обладают нормальными размерами.
Более чем у половины больных присутствуют дефекты врожденного характера: гиперпигментация кожных покровов, маленькая голова и рост, недоразвитость или полное отсутствие больных пальцев на верхних конечностях.
У некоторых пациентов с анемией Фанкони наблюдается умственная отсталость, серьезные проблемы со слухом и зрением, дефекты почек, врожденная недоразвитость половой системы, сердечные патологии.
Аномалии могут быть как сильно выраженными, так и незначительными.
Взрослея, дети с анемией Фанкони начинают все больше страдать от своего заболевания, сильно угнетающего деятельность котного мозга. При этом недуге уменьшается количество кровяных клеток.
По достижении детьми 15 летнего возраста наблюдается склонность к образованию опухолей. При анемии Фанкони увеличивается риск развития острого миелоидного лейкоза, рака печени и пищевода.
Как передается анемия Фанкони
Анемию Фанкони причисляют к наследственным болезням, которые передаются от одного поколения к другому. Тип передачи — аутосомно-рецессивный. Это означает, что ребенок заболеет лишь в том случае, если получит мутирующий ген от матери с отцом. Примечателен факт, что родители могут не болеть этим недугом, а просто являться его носителями.
При наличии аномального гена только у одного родителя малыш не заболеет, но окажется носителем анемии Фанкони (болезнь не будет себя никак проявлять), и в будущем патология может передаться его потомкам.
Иногда мальчики унаследуют недуг от матерей, если те являются носителями измененной Х-хромосомы. В этом случае у детей мужского пола риск заболеть повышается до 50%.
Лечение
Детей, у которых диагностирована анемия Фанкони, помещают на лечение в стационар гематологического отделения. Тактика терапии выстраивается исходя из тяжести протекания заболевания.
Для лечения данной патологии применяют анаболические стероиды и препараты из серии иммунно депрессантов.
Хорошие результаты в терапии анемии Фанкони дает стимуляция процессов кроветворения при помощи специальных медикаментов.
Но одной из самых эффективных терапевтических методик является трансплантация костного мозга. Проблема в том, что у этого способа имеются противопоказания, он часто сопровождается осложнениями. Наиболее подходящий возраст для хирургического вмешательства до 10 лет.
При отсутствии возможности выполнения трансплантации назначается консервативное лечение, позволяющее добиваться временного улучшения. Синтез кровных клеток стимулируется за счет андрогенов.
При наличии противопоказаний к трансплантации и стимуляции процессов кроветворения выполняют переливание составляющих крови.
Осложнения
Самые распространенные осложнения при анемии Фанкони – это частые инфекции. Дети с таким заболеванием особенно подвержены ангинам и ОРВИ. К более серьезным осложнениям относятся опухолевые новообразования. Рак у таких пациентов плохо поддается терапии. Это связано со сниженным восстановлением ДНК и чрезмерной ломкостью хромосом. Плохая свертываемость приводит к большим кровопотерям.
Пациенты с анемией Фанкони редко проживают больше 30 лет.
Профилактика
Предотвратить анемию Фанкони у ребенка можно одним единственным способом: на этапе планирования беременности не пренебрегать обследованиями Супружеским парам, готовящимся стать родителями, рекомендована медико-генетическая консультация, чтобы оценить вероятность риска рождения малыша с подобной патологией. Заболевание можно выявить и в ходе пренатальной диагностики. При обнаружении такой болезни во время беременности придется решать вопрос с ее прерыванием.
При подозрении у ребенка на анемию Фанкони нужно обратиться к врачу гематологу. Направление в таких случаях выписывает педиатр. Ребенка необходимо тщательно обследовать и, если диагноз подтвердится, разработать стратегию лечения, которой должны неукоснительно следовать родители.
Симптомы развития анемии Фанкони. Анемия фанкони у детей
Причины и факторы риска
Заболевание передается по наследству. Встречается редко — в среднем на 350 тыс. младенцев 1 ребенок рождается с анемией Фанкони; причины патологии состоят в генетической мутации.
В большинстве случаев передача заболевания происходит по аутосомно-рецессивному типу наследования. Такой способ предполагает, что ребенок может заболеть, только если оба родителя являются носителями гена с мутацией. В этом случае вероятность появления болезни у детей — 1 к 4. Родители ребенка при этом могут не быть больными.
Гораздо реже патология вызывается мутацией в гене, который сцеплен с Х-хромосомой. В таком случае женщины не заболевают, а являются носителями мутации; их сыновья в половине случаев имеют врожденную патологию.
Заболевание относится к генетическим, поэтому на вероятность появления диагноза не влияют внешние причины — в окружающей среде нет факторов риска, которые могут спровоцировать нарушение. Риск возникновения проблемы увеличивается при заключении браков между родственниками.
Симптомы и диагностика
Большинство пациентов, около 60-70%, имеют врожденные признаки заболевания, которые появляются из-за нарушения развития плода. На наличие анемии Фанкони могут указывать:
- низкий рост;
- маленькая голова;
- косоглазие;
- недостаточное развитие одного глаза или двух;
- опущение века;
- нистагм;
- нарушение развития половых органов;
- нарушение строения или функции почек;
- порок сердца;
- маленький размер большого пальца на руках или его отсутствие;
- вывих бедра;
- наличие шейного ребра;
- недостаточное развитие лучевой кости;
- косолапость;
- недоразвитость подбородка.
Могут наблюдаться нарушения в пигментации кожи — на теле бывают светлые или коричневые пятна. Иногда заболевание протекает с задержкой умственного развития.
В других случаях симптомы появляются по мере развития заболевания, т.к. возникает все больше нарушений в функционировании костного мозга. Чаще всего симптоматика начинает проявляться в детстве, в период от 5 до 10 лет.
Для больных характерна повышенная кровоточивость: можно заметить кровоподтеки на теле, при повреждении кожи кровь длительное время не сворачивается, возникают носовые кровотечения.
Ребенок может отличаться слабостью и быстрой утомляемостью, быть подверженным частым инфекционным заболеваниям. В редких случаях анемия Фанкони никак не проявляется и не диагностируется до возникновения осложнений в виде онкологических заболеваний.
Для постановки диагноза врач опрашивает больного и его семью, чтобы выяснить, встречалось ли данная патология у родственников. Проводит осмотр пациента на предмет выявления нарушений развития, которые можно увидеть невооруженным глазом. Из лабораторных исследований назначают анализы крови, генетический тест клеток и миелограмму.
Для больных анемией Фанкони при общем анализе крови характерно снижение числа эритроцитов, лейкоцитов или тромбоцитов. При этом может присутствовать недостаточность всех видов форменных элементов крови или только некоторых из них. Цитогенетическое исследование показывает наличие изменения структуры хромосом. Молекулярно-генетический анализ клеток позволяет обнаружить мутации в генах, которые отвечают за развитие анемии Фанкони. Миелограмма показывает клеточный состав костного мозга.
Лечение
Для поддержания процессов кроветворения применяется гормональная терапия. Назначают прием гемопоэтических факторов роста, иммунодепрессантов и мужских половых гормонов. Медикаменты помогают поддерживать качество жизни пациента, но со временем эффективность лечения снижается и появляются побочные эффекты. При отмене препаратов положительные изменения исчезают.
Самый эффективный метод лечения — пересадка костного мозга. Наилучшие результаты возникают при проведении операции в детском возрасте (до 10 лет) и при использовании в качестве донора здоровых братьев или сестер. При операции больший риск развития осложнений, т.к. больные анемией Фанкони из-за ломкости хромосом плохо переносят кондиционирование, необходимое для трансплантации.
Если к медикаментозной терапии и трансплантации костного мозга есть противопоказания, проводят переливание компонентов крови.
При травмах и ранениях необходимо правильно оказывать первую помощь больным с анемией из-за плохой свертываемости крови.
Профилактика
Из-за генетического фактора в возникновении заболевания его невозможно избежать, придерживаясь здорового образа жизни или соблюдая правильное питание.
Можно предварительно узнать вероятность возникновения заболевания у будущего потомства, пройдя пренатальную диагностику. Это рекомендуется делать парам, которые попадают в группу риска. Туда относятся люди, у которых встречались случаи данного заболевания в семье, и те, кто планирует родить ребенка от человека, с которым состоят в родстве.
Лечение патологии у взрослых и детей
Лечение анемии Фанкони — сложный процесс, он направлен на восстановление активности костного мозга и борьбу с осложнениями. Обязательно нужно прекратить приём лекарственных препаратов, которые могут угнетать функцию кроветворения.
На начальных этапах заболевания лечение проводят с помощью мужских половых гормонов (Неробол, Ретаболил) и препаратов, которые стимулируют выработку клеток крови (Эритропоэтин). Только в половине случаев это позволяет затормозить прогрессирование заболевания и стимулировать кроветворение.
Препараты для стимуляции костного мозга увеличивают количество красных телец в крови
Для воздействия на очаги инфекции в организме пациентов применяются антибиотики широкого спектра.
Антибиотики пациентам назначают для борьбы с инфекционными осложнениями
При тяжёлом течении анемии проводят переливания эритроцитарной и тромбоцитарной массы. Этот метод лечения позволяет увеличить содержание клеток в крови.
Переливание эритроцитарной массы улучшает снабжение тканей кислородом
Что такое эритроцитаферез и кому показана эта процедура: https://krasnayakrov.ru/donorstvo/eritrocitoferez.html
Если состояние больного средней тяжести и у него отсутствует сепсис и выраженная кровоточивость, прибегают к операции по поводу удаления селезёнки. Положительный эффект при таком методе лечения наблюдается в половине случаев. После оперативного вмешательства рекомендовано введение антилимфоцитарного иммуноглобулина. Улучшение состояния наблюдается через 3–5 месяцев, но кровоточивость заметно уменьшается уже в первые дни.
Пересадка костного мозга даёт надежду на продление жизни
Наиболее перспективным методом лечения анемии Фанкони считается пересадка генетически совместимого костного мозга. Донорами могут быть только родные братья или сёстры.
При отсутствии подходящего донора некоторые семейные пары прибегают к искусственному зачатию плода в пробирке, чтобы взять у него клетки костного мозга при условии их совместимости.
Операция трансплантации костного мозга даёт надежду на благоприятный результат, если в прошлом отсутствовали переливания крови. Необходимо проводить такое вмешательство как можно раньше. После десятилетнего возраста трансплантация костного мозга, как правило, безуспешна. Для профилактики отторжения трансплантанта назначают антилимфоцитарный гаммаглобулин и цитостатические препараты с целью подавления избыточной активности иммунной системы (Циклофосфан, Метотрексат натрия).
Метотрексат угнетает иммунные клетки
Однако следует учитывать, что большие дозы имуннодепрессантов и лучевая терапия для профилактики отторжения трансплантанта противопоказаны больным с анемией Фанкони. С этим часто связаны неудачи трансплантации.
В ситуациях, когда радикальные методы лечения не могут быть проведены, назначают кортикостероидные гормоны (Преднизолон, Дексаметазон) в индивидуальных дозировках.
Преднизолон назначается при невозможности произвести трансплантацию костного мозга
Эти препараты препятствуют развитию опухолевых процессов кроветворной системы и кровотечений.
Прогноз
Анемия Фанкони имеет относительно неблагоприятный прогноз. Консервативная терапия и трансплантация костного мозга помогают в 50% случаев. При развитии миелоидного лейкоза прогноз ухудшается.
Средняя продолжительность жизни пациентов – 30 лет. Как правило, летальный исход наступает в результате повышенной кровоточивости и присоединения оппортунистических инфекций на фоне угнетения иммунитета.
Симптомы анемии Фанкони у детей
Впервые болезнь проявляет себя в возрасте 4-10 лет, при рождении анемия Фанкони диагностируется редко.
К ее ранним симптомам относят:
-
Кровотечения, которые развиваются спонтанно.
-
Появление подкожных гематом.
-
Бледность кожи, бледность слизистых оболочек.
-
Повышенная утомляемость, вялость.
Ребенок болеет чаще своих сверстников, так как его иммунная система не работает в полную силу. Возможно развитие лимфаденопатии.
Большинство людей, страдающих анемией Фанкони, имеют врожденные дефекты развития. Они диагностируются примерно у 75% больных.
К ним относят:
-
Гиперпигментация кожных покровов с появлением характерных бронзово-коричневых пятен и пятен «кофе с молоком». К изменению цвета кожи приводит чрезмерное отложение меланина в базальном слое эпидермиса.
-
Плохое состояние зубов и ногтей.
-
Низкий рост.
-
Маленькая голова.
-
Косолапость, врожденный вывих бедра, отсутствие пальцев на руках, либо их укорочение, слабое развитие лучевой кости, шейное ребро.
-
Нарушения в умственном развитии.
-
Косоглазие, птоз.
-
Недоразвитие глаз (одного или двух).
-
Глухота.
-
Гипоплазия половых органов, монорхизм, аномальное расположение мочеиспускательного канала.
-
Патологии развития почек: наличие двух лоханок или мочеточников, недоразвитие органов, формирование почки в виде подковы, наличие кист в почках.
-
Пороки сердца.
Перечисленные нарушения развития могут присутствовать не всегда и не в полном объеме, даже если от анемии Фанкони страдают члены одной семьи. Чем старше становится человек, тем сильнее нарушаются кроветворные функции костного мозга.
В период от 15 до 25 лет у больных сохраняется высокий риск развития раковых патологий:
-
Лейкоз.
-
Карцинома языка.
-
Опухоли пищевода.
-
Опухоли печени.
Патогенез
В норме в клетках организма существуют специальные ферментные системы, которые исправляют разрывы молекул ДНК, поврежденных в процессе биосинтеза или воздействия химических, физических реагентов. При анемии Фанкони обнаруживается генетический дефект в кластере белков, ответственных за репарацию ДНК, что приводит к повышенной ломкости хромосом. В итоге у пациентов развиваются нарушения функций костного мозга – неоплазии и апластическая анемия. Онкологические заболевания чаще всего представлены острым миелоидным лейкозом – злокачественной опухолью миелоидного ростка крови, провоцирующей накопление измененных белых клеток, подавляющих рост эритроцитов, тромбоцитов и нормальных лейкоцитов. При апластической анемии в результате дисплазии костного мозга резко угнетается рост и созревание всех трех видов клеток крови.
Возможные осложнения и последствия
Наиболее частыми осложнениями анемии Фанкони являются:
- присоединение вторичной инфекции;
- повышенная кровоточивость;
- развитие лейкемии.
Онкологические центры
Клиника Ихилов
Национальный Центр Рака
Университетская клиника Шарите
Клиника Шиба (Медицинский центр Имени Хаима Шибы)
Отправить запрос
Онкологические специальности
Методы лечения
Специалисты
Профессор Арнон Наглер
Клиника Шиба (Медицинский центр Имени Хаима Шибы)Онкогематолог
Общие сведения
Синонимичные названия анемии Фанкони – врожденная панмиелопатия Фанкони, наследственная панмиелопатия. Заболевание названо по фамилии швейцарского педиатра Гвидо Фанкони, который в 1927 году описал врожденную апластическую патологию на основе симптомов у трех братьев. Анемия Фанкони является редкой генетической болезнью, наследуется согласно аутосомно-рецессивному принципу. Эпидемиологические показатели низкие – 1 больной ребенок на 350 тысяч новорожденных. Распространенность одинакова среди представителей женского и мужского пола, выше в сообществах с разрешенными близкородственными браками, например, у некоторых южноафриканских народов.
Анемия Фанкони
Причины заболевания
Причина анемии Фанкони — дефект стволовых клеток костного мозга, который возникает в результате генетической аномалии. Врождённые нарушения в группе белков, которые отвечают за восстановление молекулы ДНК, приводят к возникновению проблем с кроветворением. На данный момент известны 13 генов, изменения в которых могут вызвать эту болезнь. Анемию Фанкони может передать ребёнку отец или мать.
Причина анемии Фанкони — аномалия генов
Если ген передал только один родитель, оно клинически не проявится. Такой человек является носителем этой патологии. Признаки будут присутствовать при условии наличия у ребёнка болезненного гена от двух родителей. В таких ситуациях вероятность проявления анемии Фанкони составляет 25%.
Иногда причиной состояния бывает несовместимость крови матери и плода.
Источники
- https://MedicalOk.ru/anemiya/fankoni.html
- https://krasnayakrov.ru/organizm-cheloveka/anemiya-fankoni-u-detey.html
- https://liqmed.ru/disease/anemiya-fankoni/
- https://www.ayzdorov.ru/lechenie_anemiya_fankoni_deti.php
- https://www.KrasotaiMedicina.ru/diseases/genetic/Fanconi-anemia
- https://www.neboleem.net/anemija-fankoni.php
- https://cancer-treatment-abroad.ru/onko/gematologiya/anemiya-fankoni/
[свернуть]
Анемия Фанкони: причины, симптомы, диагностика, лечение
Лабораторные признаки анемии Фанкони
Трёхростковая аплазия выступает наиболее типичной манифестацией анемии Фанкони, однако наблюдения за инициально гематологически интактными гомозиготами показали, что зачастую тромбоцито- или лейкопения предшествуют развитию панцитопении. Первые гематологические аномалии при анемии Фанкони закономерно обнаруживают после респираторных вирусных инфекций, прививок, иногда гепатитов - так, как это характерно и для идиопатических апластических анемий. Для анемии Фанкони даже в доанемическую фазу типичен выраженный макроцитоз, сопровождающийся значительным повышением уровня фетального гемоглобина. Пунктат костного мозга, как правило, обеднён кроветворными клеточными элементами, преобладают лимфоциты, встречаются плазматические, тучные клетки и стромальные элементы - клиническая картина, неотличимая от идиопатической апластической анемии. Зачастую в аспирате костного мозга обнаруживают дисмиелопоэз и дизэритропоэз, в частности, мегалобластоидность, благодаря которой Фанкони назвал эту анемию «пернициозиформной». В биоптатах костного мозга на ранних стадиях заболевания выявляются гиперклеточные участки активного резидуального гемопоэза, которые исчезают по мере прогрессирования заболевания.
Один из фундаментальных феноменов, характерных для клеток крови больных анемией Фанкони, - это их склонность к формированию специфических хромосомных аномалий - разрывов, сестринских обменов, эндоредупликаций при культивировании клеток in vitro. Инкубация ФГА-стимулированных лимфоцитов больных анемией Фанкони с бифункциональными алкилирующими агентами, которые вызывают сшивки ДНК между гуанидиновыми основаниями, расположенными как на одной, так и на двух комплементарных цепях - нитроген-мустардом, препаратами платины, митомицином и особенно диэпоксибутаном - резко увеличивает количество аберраций. Этот феномен, получивший название кластогенного эффекта, лежит в основе современной диагностики и дифференциальной диагностики анемии Фанкони, поскольку спонтанные аберрации могут как отсутствовать у больных анемией Фанкони, так и присутствовать у больных с другими синдромами, в частности с синдромом Ниймеген. Под влиянием бифункциональных алкилирующих агентов происходит замедление клеточного цикла: клетки больных анемией Фанкони останавливаются в G2 фазе митотического цикла, что послужило основанием для разработки ещё одного диагностического теста для анемии Фанкони с помощью метода проточной флюориметрии.
Возраст первого появления анемии Фанкони в одной семье часто конкордантен, но может и существенно варьировать, в том числе и у однояйцевых близнецов. В прошлом при отсутствии специфического лечения (андрогены или трансплантация костного мозга) и проведении только гемотрансфузий заболевание неуклонно прогрессировало: 80% больных умирали от осложнений панцитопении в течение 2 лет после установления диагноза апластической анемии и практически все больные умирали через 4 года. Необходимо упомянуть, что зафиксировано несколько случаев спонтанного улучшения и даже полного восстановления гематологических показателей.
Вторыми по частоте развития гематологической презентацией анемии Фанкони выступают острые лейкозы и миелодиспластические синдромы. Примерно у 10% больных анемией Фанкони, клинические случаи которых описаны в литературе, впоследствии развился острый лейкоз. Во всех случаях, за исключением 2, лейкозы были миелоидными. Описаны даже случаи установления диагноза анемии Фанкони у пациента с резидуальной цитопенией через много лет после успешной химиотерапии ОМЛ. Несколько ниже частота развития миелодиспластических синдромов - около 5%, причём только у 1/5 из этих больных прослежена дальнейшая эволюция МДС в ОМЛ и несколько больных с МДС прожили более 10 лет. Согласно исследованиям Международного регистра анемии Фанкони риск развития ОМЛ или МДС у больных анемией Фанкони равен 52% к 40 годам. Зачастую выявляют кариотипические аномалии (моносомию 7, трисомию 21, делецию 1), которые позволяют квалифицировать ОМЛ и МДС у больных анемией Фанкони как вторичные. Интересно, что, хотя риск развития МДС/ОМЛ у больных с хромосомными аномалиями примерно в 10 раз выше, чем без таковых, наличие хромосомных аберраций не означает обязательного развития МДС. Клоны, несущие аномалии, могут спонтанно исчезать или сменять друг друга.
Кроме гематологических аномалий для больных анемией Фанкони характерна склонность к развитию опухолей. Риск развития злокачественных опухолей у больных анемией Фанкони составляет 10%, из них 5% приходится на долю опухолей печени и 5% - на остальные опухоли. Опухоли реже встречаются у детей - средний возраст диагностики опухолей печени составляет 16 лет, а остальных опухолей - 23 года. Опухоли печени (гепатоцеллюлярная карцинома, гепатома, аденома и др.), а также пелиоз («кровяные озерца») встречаются чаще у мужчин (соотношение 1,6:1), причём применение андрогенов увеличивает риск их возникновения. В то же время внепечёночные опухоли чаще встречаются у женщин (соотношение 3:1) даже после исключения опухолей гинекологической сферы. Самые частые формы рака при анемии Фанкони - чешуйчатоклеточные карциномы языка и рак пищевода, на которые приходится более 30% всех внепечёночных опухолей при анемии Фанкони, остальные опухоли встречаются в 5-7 раз реже.
Фанкони анемия | Фонд «Подари жизнь»
Суть болезни
Анемия Фанкони (АФ) – генетическое заболевание, проявляющееся в виде целого комплекса симптомов. Наиболее серьезными и опасными среди них являются развивающиеся в ходе этой болезни гематологические нарушения и опухоли.
У большинства больных анемией Фанкони в течение жизни постепенно развивается аплазия кроветворения, которая приводит к панцитопении, то есть к дефициту всех видов клеток крови. Однако, хотя АФ считается в основном заболеванием кроветворной системы, фактически при ней поражены многие органы и системы органов, причем уже при рождении часто выявляются пороки развития.
У больных АФ резко повышена вероятность возникновения в раннем возрасте гематологических и онкологических заболеваний, включая острый миелоидный лейкоз, миелодиспластические синдромы, а также опухоли пищевода, печени, желудка, верхних дыхательных путей, женских половых органов.
Иногда анемию Фанкони называют «врожденной апластической анемией», хотя проявления АФ и «обычной» (приобретенной) апластической анемии не во всем совпадают.
Частота встречаемости, факторы риска
Анемия Фанкони – редкое заболевание. Частота рождения детей с АФ в среднем составляет приблизительно 1 случай на 350 тысяч новорожденных. АФ возникает с одинаковой частотой у мужчин и у женщин и встречается среди всех национальностей. Анемия Фанкони – врожденное заболевание, которое наследуется по аутосомно-рецессивному типу. Это значит, что если и отец, и мать являются носителями дефектного гена (при этом они могут быть полностью клинически здоровыми), то при каждом деторождении есть 25% риск появления ребенка с этим заболеванием. Поэтому частота возникновения АФ повышена в сообществах людей, где приняты близкородственные браки.
Признаки и симптомы
Дети с анемией Фанкони часто обладают характерными внешними признаками. Среди возможных особенностей известны пороки развития скелета (например, больших пальцев рук, костей запястья, бедер, позвоночника, ребер), низкий рост, «птичье лицо» (недоразвитый подбородок), пятна на коже – светлые или типа «кофе с молоком». Нередки врожденные аномалии глаз, ушей, сердца, почек, мочеполовой системы. Встречается задержка физического развития. Интеллект у больных АФ, как правило, нормален.
Наиболее серьезные проблемы при АФ связаны с постепенно развивающимися нарушениями работы костного мозга. Обычно они становятся заметными с возраста 5-10 лет и затем углубляются с течением времени, хотя иногда болезнь протекает практически бессимптомно в течение многих лет.
Снижение числа тромбоцитов ведет к повышенной кровоточивости (возникают носовые кровотечения, синяки «без причины»), анемия из-за снижения числа эритроцитов – к слабости и утомляемости, а снижение числа лейкоцитов (нейтрофилов) – к плохой сопротивляемости инфекциям. Наконец, как уже говорилось, у многих больных впоследствии развивается лейкоз или миелодиспластический синдром, а также другие онкологические заболевания.
При отсутствии пороков развития и медленном нарастании симптомов болезнь порой впервые диагностируется только тогда, когда уже возникло одно из ее серьезных осложнений в виде лейкоза, миелодиспластического синдрома или другого онкологического заболевания. В этом случае прогноз ухудшается.
Диагностика
Заподозрить АФ у ребенка можно по некоторым внешним признакам, перечисленным выше, в сочетании с изменениями в клиническом анализе крови. Может наблюдаться как недостаток всех клеток крови, так и снижение числа только тромбоцитов или лейкоцитов. Для уточнения диагноза полезны исследования клеточного состава костного мозга (миелограмма). Могут использоваться и другие обследования – например, рентгенография для обнаружения и изучения врожденных дефектов костей.
Окончательный диагноз АФ можно установить после цитогенетического исследования клеток крови больного – проба с диэпоксибутаном, митомицином С или аналогичные тесты. Это связано с тем, что хромосомы клеток при АФ характеризуются определенными особенностями – повышенной нестабильностью («ломкость хромосом»). Возможен также молекулярно-генетический анализ для обнаружения конкретных генетических дефектов, однако его проведение возможно только в специальных лабораториях.
Во всех случаях снижения показателей крови (цитопении) необходимо четко отличать АФ от приобретенной апластической анемии (АА), поскольку подходы к лечению больных АА и АФ, включая тактику трансплантации костного мозга, различаются.
Семьям, где уже были случаи рождения детей с АФ, при всех последующих беременностях рекомендуется пренатальная диагностика с помощью кордоцентеза (исследования крови из пуповинной вены плода – проба с диэпоксибутаном), чтобы узнать о наличии болезни у будущего ребенка еще до его рождения. Также обследование на АФ целесообразно при рождении ребенка с аномалиями скелета, особенно кистей рук.
Лечение
Аллогенная трансплантация костного мозга - единственный шанс достичь нормализации кроветворения у больных АФ. При этом очень желательно произвести трансплантацию, пока пациент еще в молодом возрасте, оптимально - до 10 лет. Если у больного есть здоровые братья и сестры, являющиеся совместимыми потенциальными донорами для него, то предпочтительна трансплантация от них, а не от неродственного донора.
Пациенты с АФ особенно чувствительны к химиотерапии (а также лучевой терапии) и нуждаются в применении специальных протоколов кондиционирования. У них чаще обычного встречаются такие осложнения трансплантации, как токсическое воздействие терапии кондиционирования на различные внутренние органы и реакция «трансплантат против хозяина» - острая и хроническая.
Больные, для которых по тем или иным причинам невозможна трансплантация, могут получать терапию, которая временно улучшит их состояние. Так, выработку клеток крови можно стимулировать андрогенами (мужскими гормонами), а также в ряде случаев факторами роста (эритропоэтин, Г-КСФ). Примерно половина больных положительно отвечает на лечение андрогенами. Порой такая терапия помогает в течение многих лет, но со временем становится менее эффективной и связана с рядом побочных эффектов. С заместительной целью применяются также переливания компонентов крови.
Прогноз
Продолжительность жизни больных анемией Фанкони зависит от того, насколько сильно нарушена функция костного мозга. Некоторые больные живут по 30–40 лет без лечения, но многие погибают еще в детском возрасте или от самой болезни, или от развившихся в связи с ней онкологических заболеваний. Поэтому так важно вовремя провести аллогенную трансплантацию костного мозга – это единственный шанс на восстановление нормального кроветворения и на увеличение продолжительности жизни.
Трансплантация в детском возрасте от здорового совместимого родственного донора (брата или сестры) приводит к успеху у большинства больных АФ, хотя и после такой трансплантации они должны регулярно проходить осмотр для раннего выявления возможных злокачественных опухолей различных органов – вероятность их возникновения остается повышенной даже после трансплантации.
При трансплантации от неродственного донора результаты хуже, чем при использовании родственного донора, хотя и они постепенно улучшаются.
Анемия Фанкони — Википедия
Материал из Википедии — свободной энциклопедии
Анеми́я Фанко́ни — редкое наследственное заболевание. Встречается с частотой 1 на 350 000 новорождённых. Чаще выявляется среди евреев-ашкеназов и у народов Южной Африки[1].
Анемия Фанкони возникает при наличии дефекта в кластере белков, отвечающих за репарацию ДНК. Характерна повышенная ломкость хромосом. В результате этого в среднем к 40 годам у больных развивается неопластический процесс (чаще всего острый миелоидный лейкоз) и апластическая анемия.
У 60—75 % больных также встречаются врожденные дефекты, такие как низкорослость, ненормальная пигментация, маленькая голова, аномалии скелета (отсутствие или укорочение большого пальца рук, недоразвитие лучевой кости, врождённый вывих бедра, шейное ребро, косолапость). Кроме этого, определяется ряд неврологических расстройств (косоглазие, недоразвитие одного или обоих глаз, опущение века, глазное дрожание, глухота, умственная отсталость), поражения половых органов (недоразвитие половых органов, отсутствие одного или обоих яичек, гипоспадия), почечные аномалии (недоразвитие почек, удвоение лоханки или мочеточника, подковообразная почка, множественные кисты в тканях почек), врождённые пороки сердца. Средняя продолжительность жизни у больных анемией Фанкони составляет около 30 лет.
Анемия Фанкони в большинстве случаев имеет аутосомно-рецессивный тип наследования. Это означает, что две копии гена (одна от матери и одна от отца) несут в себе мутации. При этом вероятность рождения больного ребенка у таких родителей составляет 25 %. В редких случаях анемия Фанкони имеет Х-сцепленный рецессивный тип наследования. При этом только мать является носителем мутации, а вероятность заболевания у сыновей составляет 50 %.
В настоящее время известно 15 генов, связанных с анемией Фанкони. Только один из них — FANCB — находится на Х-хромосоме. Остальные гены расположены на аутосомах. Каждый из этих генов отвечает за синтез определённого фермента. Клетки от двух больных, выращенные в клеточной культуре в лабораторных условиях, могут дополнять друг друга и функционировать нормально, если они несут нарушения в разных генах. При наличии мутаций в одном и том же гене этого не происходит, и клетки не дополняют друг друга. По этому принципу выделяют группы комплиментации: A, B, C, D…, каждой из которых соответствует определенный ген: FANCA, FANCB, FANCC, FANCD1 (BRCA2), FANCD2, FANCE, FANCF, FANCG, FANCI, FANCJ, FANCL, FANCM, FANCN, FANCP и RAD51C. В каждой этнической группе есть мутации, характерные для неё. Так среди ашкеназов чаще всего встречается мутация в гене FANCC.
Для лечения применяются андрогены и гематопоэтические факторы роста, но только у 50—75 % пациентов при этом наступает улучшение, исчезающее при отмене препаратов. При значительной анемии проводят переливание крови. Известны случаи успешной пересадки костного мозга. При этом из-за повышенной ломкости хромосом для этих больных опасно облучение, применяемое перед пересадкой.
Анемия Фанкони: симптомы, диагностика, лечение, причины
Содержание статьи:
Анемия Фанкони – редкое заболевание, развитие которого обусловлено генетическим дефектом группы белков, отвечающих за репарацию (исправление разрывов и ошибок) молекул ДНК. Это приводит к повышенной ломкости хромосом, в результате чего у пациентов с возрастом развивается апластическая анемия и запускаются неопластические процессы.
Анемия Фанкони встречается с частотой 1 случая на 350 000 новорожденных. Более высокий показатель заболеваемости отмечается у жителей Южной Африки и евреев-ашкенази – 1 случай на 90 новорожденных.
Внимание! Фотография шокирующего содержания.
Для просмотра нажмите на ссылку.
Причины и факторы риска
В большинстве случаев для анемии Фанкони характерен аутосомно-рецессивный тип наследования. Это означает, что для развития патологии ребенок должен получить мутировавший ген и от матери, и от отца. В тех случаях, когда он получает только одну копию измененного гена, заболевание не разовьется, но ребенок будет носителем патологического гена и сможет в дальнейшем передать его своим потомкам. Если и отец, и мать являются носителями мутированного гена, вероятность рождения больного ребенка – 25%.
Существует форма анемии Фанкони с рецессивным Х-сцепленным типом наследования. Носители мутации в этом случае – женщины. Вероятность рождения у них сына с анемией Фанкони составляет 50%.
 Анемия Фанкони наследуется по аутосомно-рецессивному типу – ребенок должен получить мутированных ген от обоих родителей
Анемия Фанкони наследуется по аутосомно-рецессивному типу – ребенок должен получить мутированных ген от обоих родителей Симптомы
Первые признаки нарушения кроветворения при анемии Фанкони начинают проявляться у детей после 6-7 лет. Но заподозрить данную патологию можно и ранее по внешнему облику пациентов, для которых характерны:
- низкорослость;
- микрофтальмия;
- микроцефалия;
- смуглая кожа;
- неправильно сформированные большие пальцы рук;
- наличие на кожных покровах и слизистых оболочках участков гипер- и гипопигментации.
Прогноз при анемии Фанкони серьезный. Большинство больных живут не более 40 лет.
Помимо этого, могут наблюдаться другие аномалии:
- умственная отсталость;
- глухота;
- глазное дрожание;
- косоглазие;
- птоз века;
- гипоспадия;
- отсутствие яичек, недоразвитие половых органов;
- врожденные пороки сердца;
- поликистоз или гипоплазия почек, подковообразные почки;
- удвоение мочеточников и (или) лоханок.
При анемии Фанкони постепенно нарушается кроветворная функция костного мозга. В первые годы жизни уровень клеток крови обычно соответствует нормальным возрастным показателям, но эритроциты отличаются значительными размерами (макроцитоз). В последующие годы у пациентов развивается панцитопения, то есть уровень всех линий клеток крови снижается.
Уменьшение количества тромбоцитов становится причиной появления кровоподтеков, частых носовых кровотечений. В результате снижения числа эритроцитов возникает анемия с такими характерными симптомами:
- бледность кожи;
- слабость;
- тахикардия;
- чувство нехватки воздуха, одышка.
Уменьшение количества нейтрофилов (нейтропения) приводит к низкой сопротивляемости организма инфекциям.
Трансплантация донорского костного мозга – единственная возможность добиться восстановления функции кроветворения при анемии Фанкони.
В дальнейшем нередко развиваются онкологические заболевания (в частности, лейкозы и миелодиспластический синдром).
Читайте также:Польза и вред сдачи крови: 12 заблуждений о донорстве
8 продуктов, помогающих улучшить кровообращение
Целебная гречка: 8 свойств знакомого продукта
Диагностика
Предположить наличие анемии Фанкони можно на основании характерного внешнего вида пациента и изменений в клиническом анализе крови. Уточнить диагноз позволяет миелограмма (анализ клеточного состава костного мозга).
Для постановки окончательного диагноза необходимо цитогенетическое исследование клеток крови (тест с митомицином С или проба с диэпоксибутаном). В специализированных лабораториях может быть выполнен и молекулярно-генетический анализ, позволяющий выявить у пациента наличие конкретного генетического дефекта.
Лечение
Единственным методом, позволяющим добиться восстановления кроветворения при анемии Фанкони, является трансплантация донорского костного мозга. Желательно осуществить ее в первое десятилетие жизни ребенка, так как в старшем возрасте эффективность трансплантации значительно снижается. Наилучшие результаты наблюдаются в тех случаях, когда в качестве доноров костного мозга выступают близкие родственники больного, не имеющие данного заболевания.
При подготовке пациентов к пересадке костного мозга при анемии Фанкони нужно учитывать следующие особенности: они обладают повышенной чувствительностью к повреждающему воздействию химиотерапевтических препаратов и ионизирующего излучения, у них повышен риск развития хронических и острых реакций «трансплантат против хозяина».
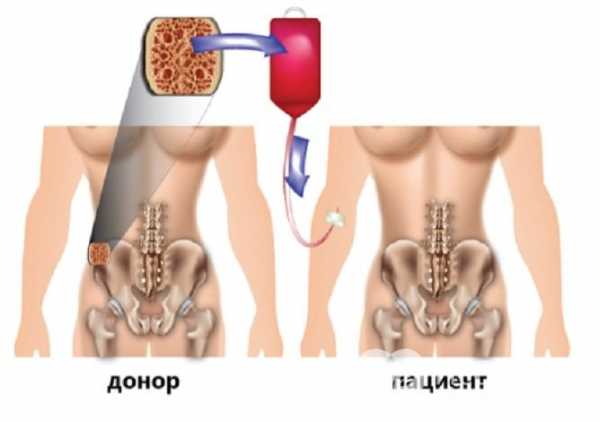 При анемии Фанкони показана пересадка костного мозга от донора
При анемии Фанкони показана пересадка костного мозга от донора Если в силу каких-либо причин трансплантация костного мозга невозможна, проводят консервативную терапию, направленную на стимуляцию процессов гемопоэза. С этой целью назначают лекарственные препараты мужских половых гормонов (андрогенов) или факторов роста. Примерно в 50% случаев обеспечивается хороший ответ на гормональную терапию андрогенами, и в течение многих лет она позволяет поддерживать выработку клеток крови костным мозгом на должном уровне. Однако в дальнейшем ее эффективность снижается, увеличивается вероятность развития побочных эффектов.
В тяжелых случаях осуществляют переливание эритроцитарной, лейкоцитарной или тромбоцитарной массы.
Возможные осложнения и последствия
Наиболее частыми осложнениями анемии Фанкони являются:
- присоединение вторичной инфекции;
- повышенная кровоточивость;
- развитие лейкемии.
Прогноз
Прогноз при анемии Фанкони серьезный. Большинство больных живут не более 40 лет. Даже после удачно выполненной трансплантации костного мозга и восстановления функции кроветворения вероятность развития онкологических заболеваний остается очень высокой.
Если и отец, и мать являются носителями мутированного гена, вероятность рождения ребенка с анемией Фанкони – 25%.
Профилактика
Предотвратить развитие анемии Фанкони невозможно. Если в роду имеются случаи данного заболевания, беременной женщине рекомендуется пренатальная диагностика: из пуповинной вены плода берется кровь (кордоцентез), ставится проба с диэпоксибутаном. При положительном результате семейной паре предлагается провести прерывание беременности по медицинским показаниям.