Аномалия арнольда киари 1 степени что это
сколько живут, степени инвалидности, продолжительность жизни
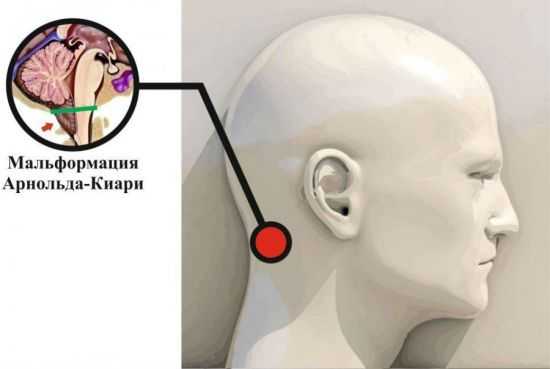
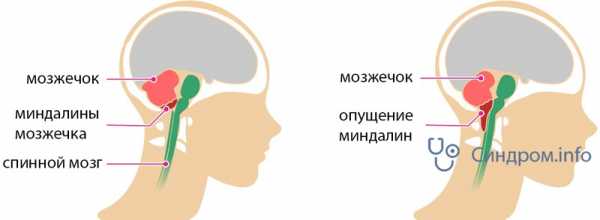
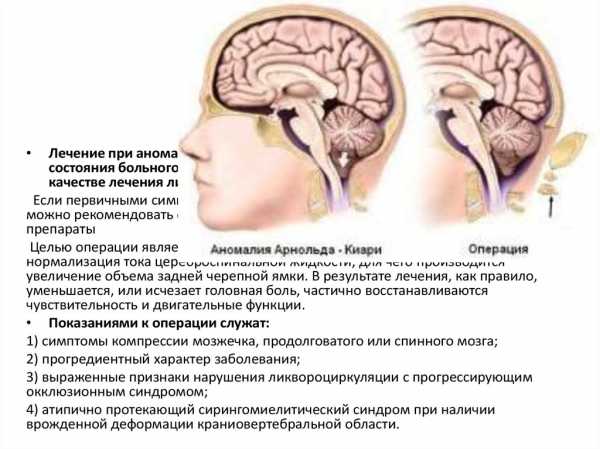
Аномалия Арнольда Киари – нарушение развития, которое заключается в виде несоразмерности размеров черепной ямки и структурных элементов мозга, располагающихся в ней. При этом мозжечковые миндалины спускаются ниже анатомического уровня и могут ущемляться.
Симптомы аномалии Арнольда Киари проявляются в виде частых головокружений, а иногда заканчиваются инсультом мозга. Признаки аномалии могут долго отсутствовать, а затем резко заявить о себе, например, после вирусной инфекции, удара головой или других провоцирующих факторов. Причем случиться это может на любом отрезке жизни.
Описание болезни
Сущность патологии сводится к неправильной локализации продолговатого мозга и мозжечка, в результате чего появляются краниоспинальные синдромы, которые врачи нередко расценивают как атипичный вариант сирингомиелии, рассеянного склероза, спинномозговой опухоли. У большинства больных аномалия развития ромбэнцефалона совмещается с другими нарушениями в спинном мозге – кистами, провоцирующими стремительную деструкцию спинномозговых структур.
Болезнь получила название в честь патологоанатома Арнольда Джулиуса (Германия), который описал аномальное отклонение в конце 18 века и врача из Австрии Ганса Киари, который изучал заболевание в тот же период времени. Распространенность нарушения варьируется в пределах 3–8 случаев на каждые 100000 человек. В основном встречается аномалия Арнольда Киари 1 и 2 степени, а взрослые с 3-м и 4-м типом аномалии живут совсем недолго.
Аномалия Арнольда Киари 1 типа заключается в опускании элементов задней черепной ямки в спинальный канал. Болезнь Киари 2 типа характеризуется изменением местоположения продолговатого мозга и четвертого желудочка, при этом зачастую бывает водянка. Гораздо реже встречается третья степень патологии, которой присущи выраженные смещения всех элементов черепной ямки. Четвертый тип представляет собой дисплазию мозжечка без его сдвига вниз.
Причины заболевания
По данным ряда авторов, болезнь Киари представляет собой недоразвитие мозжечка, сочетающееся с различными отклонениями в отделах мозга. Аномалия Арнольда Киари 1 степени – наиболее распространенная форма. Это нарушение представляет собой одностороннее или двухстороннее опускание миндалин мозжечка в спинальный канал. Это может произойти вследствие перемещения продолговатого мозга вниз, часто патология сопровождается различными нарушениями краниовертебральной границы.
Клинические проявления могут возникнуть только на 3–4 десятке жизни. При этом следует отметить, что бессимптомное течение эктопии миндалин мозжечка в лечении не нуждается и часто проявляется случайно на МРТ. На сегодняшний день этиология болезни, так же как и патогенез, изучены плохо. Определенная роль отводится генетическому фактору.
Выделяют три звена в механизме развития:
- генетически обусловленная врожденная остеоневропатия;
- травматизация ската во время родов;
- высокое давление ликвора на стенки спинномозгового канала.
Болезнь может быть вызвана наследственными нарушениями
Проявления
По частоте возникновения выделяют следующие симптомы:
- головные боли – у трети пациентов;
- боль в конечностях – 11%;
- слабость в руках и ногах (в одной или двух конечностях) – больше половины пациентов;
- чувство онемения в конечности – половина больных;
- снижение или утрата температурной и болевой восприимчивости – 40%;
- шаткость походки – 40%;
- непроизвольные колебания глаз – треть больных;
- двоение в глазах – 13%;
- нарушения глотания – 8%;
- рвота – у 5%;
- нарушения произношения – 4%;
- головокружения, глухота, онемение в лицевой области – у 3% больных;
- синкопальные (обморочные) состояния – 2%.
Боли в голове и области шеи – распространенный симптом патологии
Болезнь Киари второй степени (диагностируется у детей) сочетает в себе дислокацию мозжечка, ствола и четвертого желудочка. Неотъемлемый признак – наличие менингомиелоцеле в области поясницы (грыжа спинального канала с выпячиванием вещества спинного мозга). Неврологическая симптоматика развивается на фоне аномального строения затылочной кости и шейного отдела позвоночного столба. Во всех случаях присутствует гидроцефалия, часто – сужение водопровода мозга. Неврологические признаки появляются с самого рождения.
Операция при менингомиелоцеле проводится в первые дни после рождения. Последующее хирургическое расширение задней черепной ямки позволяет добиться хороших результатов. Многие пациенты нуждаются в шунтировании, особенно при стенозе Сильвиевого водопровода. При аномалии третьей степени черепно-мозговая грыжа внизу затылка или в верхней шейной области сочетается с нарушениями развития мозгового ствола, краниального основания и верхних позвонков шеи. Образование захватывает мозжечок и в 50% случаев – затылочную долю.
Эта патология встречается очень редко, имеет неблагоприятный прогноз и резко сокращает продолжительность жизни даже после операции. Сколько именно человек будет жить после своевременного вмешательства, точно сказать нельзя, но, вероятнее всего, что недолго, так эта патология считается несовместимой с жизнью. Четвертая степень заболевания представляет собой обособленную гипоплазию мозжечка и на сегодняшний день не относится к симптомокомплексам Арнольда-Киари.
Клинические проявления при первом типе прогрессируют медленно, в течение нескольких лет и сопровождаются включением в процесс верхнего шейного спинномозгового отдела и дистального отдела продолговатого мозга с нарушением работы мозжечка и каудальной группы черепных нервов. Таким образом, у лиц с аномалией Арнольда-Киари выделяют три неврологических синдрома:
- Бульбарный синдром сопровождается дисфункцией тройничного, лицевого, преддверно-улиткового, подъязычного и вагусного нервов. При этом наблюдаются нарушения глотания и речи, бьющий вниз спонтанный нистагм, головокружения, расстройства дыхания, парез мягкого неба с одной стороны, охриплость голоса, атаксии, дискоординация движений, неполный паралич нижних конечностей.
- Сирингомиелитический синдром проявляется атрофией мышц языка, нарушением глотания, отсутствием чувствительности в лицевой области, хриплостью голоса, нистагмом, слабостью в руках и ногах, спастическим повышением мышечного тонуса и т. д.
- Пирамидный синдром характеризуется незначительным спастическим парезом всех конечностей с гипотонусом рук и ног. Сухожильные рефлексы на конечностях повышаются, брюшные рефлексы не вызываются или снижаются.
Операция проводится при тяжелых формах нарушения
Боли в области затылка и шеи могут усиливаться при покашливании, чихании. В руках снижается температурная и болевая чувствительность, а также мышечная сила. Часто возникают обмороки, головокружения, у больных ухудшается зрение. При запущенной форме появляются апноэ (кратковременная остановка дыхания), быстрые неконтролируемые движения глаз, ухудшение глоточного рефлекса.
Интересный клинический признак у таких людей – провоцирование симптомов (синкопе, парестезии, боли и др.) натуживанием, смехом, кашлем, пробой Вальсальвы (усиленный выдох при закрытом носе и рте). При нарастании очаговых симптомов (стволовых, мозжечковых, спинномозговых) и гидроцефалии встает вопрос о хирургическом расширении задней черепной ямки (субокципитальной декомпрессии).
Диагностика
Диагноз аномалии первого типа не сопровождается повреждением спинного мозга и ставится в основном у взрослых посредством КТ и МРТ. По данным патологоанатомического вскрытия, у детей с грыжей спинномозгового канала болезнь Киари второго типа выявляют в большинстве случаев (96–100%). С помощью УЗИ можно определить нарушения циркуляции ликвора. В норме цереброспинальная жидкость легко циркулирует в подпаутинном пространстве.
Смещение миндалин вниз затрудняет циркуляцию. Из-за нарушения ликвородинамики возникает гидроцефалия.
Боковой рентген и МР картина черепа отображает расширение канала позвоночного столба на уровне С1 и С2. На ангиографии сонных артерий наблюдается огибание миндалины мозжечковой артерией. На рентгене отмечаются такие сопутствующие изменения краниовертебральной области, как недоразвитие атланта, зубовидного отростка эпистрофея, укорачивание атлантозатылочной дистанции.
При сирингомиелии на боковом снимке рентгена наблюдается недоразвитие задней дуги атланта, недоразвитие второго шейного позвонка, деформация большого затылочного отверстия, гипоплазия боковых частей атланта, расширение позвоночного канала на уровне С1-С2. Дополнительно следует провести МРТ и инвазивное рентгенологическое исследование.
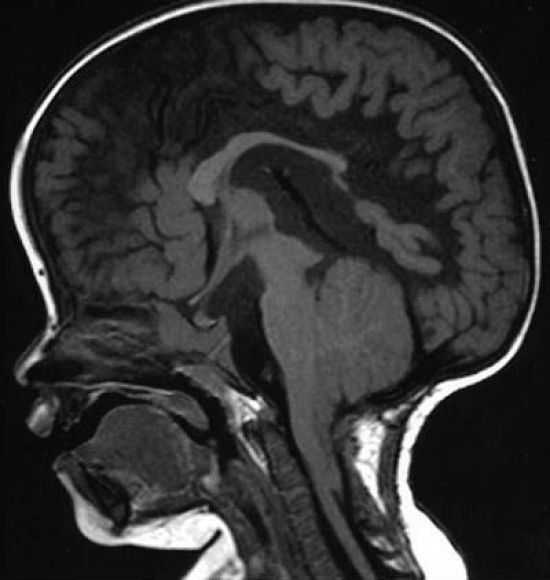
МРТ – наиболее предпочтительный способ диагностики
Манифестация симптомов болезни у взрослых и лиц пожилого возраста часто становится поводом для выявления опухолей задней черепной ямки или краниоспинальной области. В некоторых случаях правильно поставить диагноз помогают имеющиеся у пациентов внешние проявления: низкая линия оволосения, укороченная шея и др., а также наличие на рентгене, КТ и МРТ краниоспинальных признаков костных изменений.
Сегодня «золотым стандартом» диагностики нарушения является МРТ мозга и шейно-грудного отдела. Возможно внутриутробное проведение УЗИ диагностики. К вероятным ЭХО-признакам нарушения относятся внутренняя водянка, лимоноподобная форма головы и мозжечок в виде банана. В то же время некоторые специалисты не считают такие проявления специфичными.
Для уточнения диагноза используют различные плоскости сканирования, благодаря чему можно обнаружить несколько информативных в отношении болезни симптомов у плода. Получить изображение во время беременности достаточно легко. Ввиду этого УЗИ остается одним из основных вариантов сканирования для исключения патологии у плода во втором и третьем триместрах.
Выявление признаков заболевания у плода может стать показанием к исключению пороков развития позвоночного столба, однако даже абсолютное отсутствие данных УЗИ при этом указывает на расщепление позвоночника в 95% случаев.
Лечение
При бессимптомном течении показано постоянное наблюдение с регулярным ультразвуковым и рентгенографическим исследованием. Если единственный признак аномалии – незначительные боли, пациенту назначают консервативное лечение. Оно включает разнообразные варианты с использованием нестероидных противовоспалительных средств и миорелаксантов. К наиболее распространенным НПВС относятся Ибупрофен и Диклофенак.
Нельзя самостоятельно назначать себе обезболивающие препараты, так как они имеют ряд противопоказаний (например, язвенная болезнь). При наличии какого-либо противопоказания врач подберет альтернативный вариант лечения. Время от времени назначают дегидратационную терапию. Если в течение двух-трех месяцев эффекта от такого лечения нет, проводят операцию (расширение затылочного отверстия, удаление дужки позвонка и т. д.). В этом случае требуется строго индивидуальный подход, позволяющий избежать как ненужного вмешательства, так и проволочки с операцией.
Тактика лечения в отношении каждого пациента требует индивидуального подхода
У некоторых пациентов хирургическая ревизия является способом постановки конечного диагноза. Цель вмешательства – ликвидация сдавливания нервных структур и нормализация ликвородинамики. Такое лечение приводит к существенному улучшению у двух-трех пациентов. Расширение черепной ямки способствует исчезновению головных болей, восстановлению осязаемости и подвижности.
Благоприятный прогностический признак – расположение мозжечка выше С1 позвонка и наличие только мозжечковой симптоматики. В течение трех лет после вмешательства могут возникать рецидивы. Таким пациентам по решению медико-социальной комиссии присваивается инвалидность.
Мальформация Арнольда — Киари — Википедия
Мальформация Арнольда — Киари — опущение миндалин мозжечка в большое затылочное отверстие со сдавливанием продолговатого мозга. В тяжелых случаях (мальформация Киари 2) отмечаются также гидроцефалия, сирингомиелия и менингомиелоцеле. Заболевание проявляется симптомами поражения продолговатого мозга, мозжечка (затылочные боли, нарушение глотания, атаксия) разной выраженности, симптомами поражения спинного мозга и др. Может сочетаться с базилярной импрессией или инвагинацией, ассимиляцией атланта.
В норме миндалины мозжечка расположены выше большого затылочного отверстия. У пациентов с аномалией Арнольда-Киари миндалины мозжечка смещаются вниз до уровня первого, а иногда, и второго шейных позвонков, блокируя ток спинномозговой жидкости.
Ранее считалось, что аномалия Арнольда-Киари всегда носит врожденный характер, однако сейчас полагают, что у большинства людей смещение миндалин мозжечка происходит во время бурного роста мозга в условиях медленно растущих костей черепа. Только небольшое количество пациентов с аномалией Арнольда-Киари действительно имеют врожденный характер заболевания. Так же существуют другие врожденные заболевания, которые могут приводить к смещению миндалин мозжечка. К ним относятся — платибазия, базилярная инвагинация, аномалия Денди-Уокера и др.
C 2005 года существует новая теория, которая считает, что причиной Мальформации или Синдрома Арнольда Киари 1 является анормальное натяжение спинного мозга из-за напряженной концевой нити. Также эта теория связывает с натяжением спинного мозга и другие проблемы, которые часто появляются вместе с Синдромом Арнольда Киари 1: идиопатическую сирингомиелию, идиопатический сколиоз, платибазию, базиллярную импрессию и т. д.
Частота этого заболевания составляет от 3.3 до 8.2 наблюдений на 100000 населения.
Сирингомиелия развивается у 80 % больных этим заболеванием.
Средний возраст пациентов — 25-40 лет.
Типы Синдрома Арнольда Киари (САК)[1]:
САК.I. Опущение миндалин мозжечка (ОММ) без какой-либо иной мальформации нервной системы.
САК.II. ОММ с нейропозвоночной мальформацией, при которой спинной мозг прикреплен к позвоночному каналу.
САК.III. ОММ с затылочной энцефалоцеле и мозговыми аномалиями при САК.II.
САК.IV. ОММ аплазия или гипоплазия мозжечка, связанная с аплазией намёта мозжечка.
САК.0. В настоящее время зарегистрированы случаи клинической картины, свойственной САК.I, без ОММ.
САК.1.5. Недавно описан САК.1,5 с ОММ и опущением ствола головного мозга в затылочное отверстие.
На основании анализа клинико-рентгенологических и нейровизуализационных наблюдений выделено 3 варианта САК I: передний, промежуточный и задний (С. В. Можаев и соавт., Институт мозга человека РАН, Санкт-Петербург, кафедра неврологии и нейрохирургии СПбГМУ, 2007)[2]:
- передний вариант сочетает в себе отклонение зуба С2 позвонка кзади, платибазию или базилярную импрессию, а также нависание продолговатого мозга над зубовидным отростком;
- промежуточный вариант предполагает элементы компрессии вентральных отделов продолговатого и верхнешейных сегментов спинного мозга зубовидным отростком С2 позвонка и дорсальных — сместившимися миндалинами мозжечка;
- задний вариант предполагает элементы компрессии дорсальных отделов продолговатого и верхнешейных сегментов спинного мозга смещенными в большое затылочное отверстие миндалинами мозжечка.
Подзатылочная декомпрессионная краниотомия (ПДК) не устраняет причину заболевания. ПДК при Синдроме Арнольда Киари I (САК.I) всего лишь освобождает от давления на нервную систему в затылочном отверстии, что может иногда сопровождаться временными клиническими улучшениями в постоперационный период.
Данное лечение имеет высокий показатель осложнений и смертности (0,7-12 %), который можно допустить в случаях более высокой смертности, например, опухоли, сосудистых мальформаций или гематом затылочного отверстия. Но показатель внезапной смертности от заболевания, САК.I, несоизмеримо ниже показателя смертности от предлагаемого лечения, в последние три десятилетия было опубликовано 8 таких случаев:
«… in the literature, mortality rates vary from 0,7 % (AGHAKHANI, J.N. et al. 1999) to 1,4 % (PAUL, K.S. et al. 1983) and 12,1 % (LORENZO, D.N. et al. 1982) two patients of our series died in the early postoperative period, indicating a surgical mortality of 1 %…» (KLEKAMP, J.; SAMI, M. 2012).
Рассечение концевой нити при опущении миндалин мозжечка или САК.I гораздо менее травматичен, чем традиционное лечение (открытие задней черепной ямки при ПДК). Индекс осложнений и смертности при рассечении концевой нити с помощью минимально инвазивной техники равен нулю.
В расписании болезней, освобождающих от службы в армии (Постановление Правительства РФ от 04.07.2013 N 565 (ред. от 21.04.2018)), мальформация Арнольда-Киари отсутствует.
- Неврология и нейрохирургия: учебник: в 2 т./Е. И. Гусев , А. Н. Коновалов, В. И. Скворцова. — 2-е изд., испр. и доп. — М.: ГЭОТАР-Медиа, 2010. — Т.1: Неврология. — 624 с.:ил.
- ROYO, M. (1992) «Aportación a la etiología de la siringomielia», Tesis doctoral (PDF). UNIVERSIDAD AUTÓNOMA DE BARCELONA.
- ROYO, M. (1996). «Siringomielia, escoliosis y malformación de Arnold-Chiari idiopáticas, etiología común» (PDF). REV NEUROL (Barc) 1996; 24 (132): 937—959.
- ROYO, M. (1996). «Platibasia, impresión basilar, retroceso odontoideo y kinking del tronco cerebral, etiología común con la siringomielia, escoliosis y malformación de Arnold-Chiari idiopáticas» (PDF). REV NEUROL (Barc) 1996; 24 (134): 1241—1250.
- ROYO, M. (1997). «Nuevo tratamiento quirúrgico para la siringomielia, la escoliosis, la malformación de Arnold-Chiari, el kinking del tronco cerebral, el retroceso odontoideo, la impresión basilar y la platibasia idiopáticas» (PDF). REV NEUROL 1997; 25 (140): 523—530.
- KLEKAMP, J., SAMII, M., «Syringomyelia». Spinger-Verlang 2002, pag. 65.
- AGHAKHANI, J., PARKER, F., TADIE, M. (1999). «Syringomyelia and Chiari abnormality in the adult. Analysis of the results of a comparative series of 285 cases». Neurochirurgie 45 (Suppl. 1): 23-36.
- LORENZO, D.N., FORTUNA, A., GUIDETTI, B. (1982) "Craneovertebral junction malformations. Cranioradiological findings, long term results and surgical indications in 63 cases. J. Neurorusurg 57: 603—608.
- PAUL, K.S., LYE, R.H., STRANG, F.A., DUTTON, J. (1983). "Arnold-Chiari Malformation. Review of 71 cases. J. Neurosurg 58:183-187.
- ROYO, M., SOLE-LLENAS, J., DOMENECH, J.M., GONZÁLEZ-ADRIO, R. (2005). «[1]». Acta Neurochir (Wien). 2005 Feb 24.
- РЕУТОВ А. А., КАРНАУХОВ В. В. (2015). «КЛИНИЧЕСКИЕ РЕКОМЕНДАЦИИ „ХИРУРГИЧЕСКОЕ ЛЕЧЕНИЕ МАЛЬФОРМАЦИИ КИАРИ У ВЗРОСЛЫХ“». Пленум Правления Ассоциации нейрохирургов России (Санкт-Петербург). 2015 Apr 16 (PDF).
что это, причины, степени и типы, лечение
Синдром Арнольда-Киари — врожденная патология краниовертебральной зоны, возникающая в период эмбриогенеза и характеризующаяся деформацией черепа. Опущение структур головного мозга в большое затылочное отверстие приводит к их сдавлению и ущемлению. Дисфункция мозжечка сопровождается нистагмом, шаткой походкой, дискоординацией движений, а поражение продолговатого мозга — изменениями в работе жизненно важных органов и систем.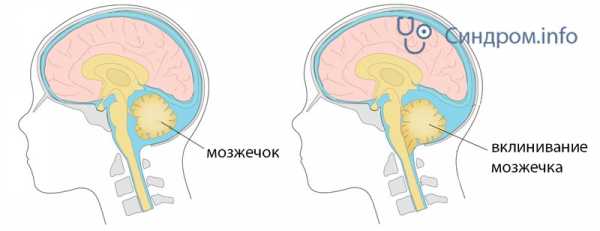
Впервые синдром был описан в конце 18 века двумя учеными: доктором из Германии Арнольдом Джулиусом и врачом из Австрии Гансом Киари. Аномалию обнаруживают сразу после появления ребенка на свет или несколько позже — в пубертатном или зрелом возрасте. Это зависит от типа синдрома. У взрослых пациентов недуг чаще всего становится неожиданной находкой. Средний возраст больных — 25-40 лет.
Симптоматика патологии также определяется типом мальформации. Хоть синдром и считается врожденным пороком, не всегда его клинические признаки возникают с самого рождения. Иногда они обнаруживаются после 40 лет. У больных возникает цефалгия, напряженность мышц шеи, головокружение, нистагм, обмороки, атаксия, нарушения речи, парез гортани, тугоухость, падение остроты зрения, нарушение процесса глотания, стридор, парестезии, слабость мышц. У большинства пациентов симптомы заболевания полностью отсутствуют. Его обнаруживают случайно, во время комплексного диагностического обследования, проводимого совсем по другому поводу. Пациентам с бессимптомными формами не требуется лечение.
При постановке диагноза учитывают данные осмотра больного и его неврологического статуса, а также результаты томографического исследования мозга. Магнитно-ядерный резонанс позволяет точно и быстро определить наличие трансформации, уровень поражения и степень ущемления головного мозга. Лечение синдрома медикаментозное, физиотерапевтическое и оперативное. Больным выполняют операции по шунтированию мозга и декомпрессии краниовертебральной зоны.
Этиология
Причины синдрома в настоящее время точно не определены. Существует несколько теорий происхождения патологии, но до конца ни одна из них не имеет официального подтверждения. Мнения ученых-неврологов всего мира до сих пор расходятся.
Большинство медиков признают синдром врожденным недугом, сформированным в процессе эмбриогенеза под воздействием негативных факторов внешней среды, оказывающих свое пагубное действие на женский организм при беременности. К ним относятся: самостоятельные использование лекарств, алкоголизм, табакокурение, вирусные инфекции, ионизирующее излучение.
Врожденные причины:
- Дистопия мозжечковых миндалин за пределы задней черепной ямки, имеющей относительно малые размеры;
- Выталкивание растущих структур мозга через затылочное отверстие;
- Неправильное формирование в процессе эмбриогенеза и атипичное развитие в постнатальном периоде костной ткани, приводящее к деформации черепной коробки.
Некоторые ученые определенную роль в развитии синдрома отводят генетическому фактору. В настоящее время можно точно утверждать, что болезнь не связана с хромосомными аномалиями.
Другие ученые придерживаются иной точки зрения относительно происхождения синдрома. Они считают его приобретенным и объясняют свое мнение появлением симптомов патологии у взрослых лиц. Приобретенный синдром возникает под действием экзогенных факторов. У больного новорожденного ребенка череп может иметь нормальное строение без костных аномалий и гипоплазии.
Приобретенные причины:
- Родовой травматизм с поражением черепа и мозга,
- Воздействие цереброспинальной жидкости на стенки спинного мозга,
- Любые ЧМТ,
- Бурный рост мозга в условиях медленно растущих костей черепа.
Симптомы патологии долгое время отсутствуют у больных, а затем внезапно появляются под воздействием провоцирующих факторов: вирусов, травм головы, стрессов.
Опущение основных мозговых структур до шейных позвонков блокирует процесс перетекания ликвора из подпаутинного пространства в спинномозговой канал. Это приводит к дисциркуляторным изменениям. Ликвор, продолжая синтезироваться и никуда не оттекая, скапливается в головном мозге.
Ученый Киари в 1891 году выделил четыре типа аномалии:
- I – выход структурных элементов головного мозга за пределы задней черепной ямки, обусловленный недоразвитием костной ткани этой области. Этот тип клинически проявляется у лиц зрелого возраста.
выход структур мозжечка за пределы ЗЧЯ при аномалия 1 типа
- II — нарушения эмбриогенеза, приводящие к расположению структур мозжечка и продолговатого мозга ниже большого затылочного отверстия.
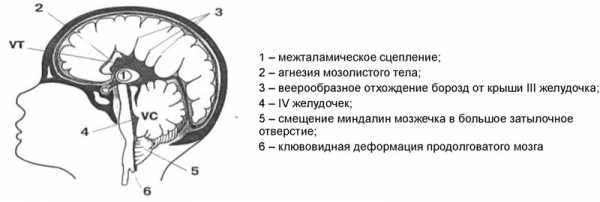
синдром Арнольда-Киари II типа
- III — эктопия мозговых структур в каудальном направлении с образованием энцефаломенингоцеле.
аномалия III типа
- IV — недоразвитый мозжечок не смещается и не выходит за пределы черепа. Поскольку отсутствует грыжевое выпячивание мозга, этот тип синдрома отсутствует в современной классификации.
Существуют два новых типа синдрома. Тип 0 — мозжечок располагается достаточно низко, но при этом находится в черепной коробке. Тип 1.5 – промежуточная форма, сочетающая признаки I и II типов.
Выделяют три степени тяжести патологии:
- Первая — относительно легкая форма патологии без аномалий мозговых структур и характерных клинических проявлений.
- Вторая — наличие пороков развития ЦНС с врожденным недоразвитием головного мозга и подкорки.
- Третья — аномалии строения головного мозга со смещением мягких тканей в отношении твердых структур, образованием ликворных кист и сглаженностью извилин.
Симптоматика

Аномалия Киари I типа — самая распространенная форма синдрома, клинические признаки которой условно объединены в пять синдромов:
- Гипертензионный синдром проявляется цефалгией, подъемом артериального давления в утренние часы, напряженностью и гипертонусом шейных мышц, дискомфортом и болезненными ощущениями в шейном отделе позвоночника, диспепсическими явлениями, общей астенизацией организма У новорожденных детей возникает общее беспокойство, рвота фонтаном, тремор подбородка и конечностей, нарушается сон. Ребенок постоянно плачет, отказывается от груди.
- При наличии мозжечковых нарушений у больных изменяется произношение, речь становится скандированной, возникает вертикальный нистагм. Они жалуются на частые головокружения, рассогласованность движений, шаткость походки, дрожание рук, нарушение равновесия, дезориентацию в пространстве. Больные с большим трудом выполняют простые целенаправленные действия, в движениях отсутствует четкость и скоординированность.
- Поражение черепно-мозговых нервов проявляется признаками корешкового синдрома. У пациентов ограничивается подвижность языка и мягкого неба, что приводит к нарушению речи и проглатывания пищи. Их голос изменяется в сторону гнусавости и осиплости, речь становится неясной, дыхание затрудненным. Нарушение ночного дыхания отмечаются у большинства больных. У них возникает гипопноэ, центральное или обструктивное апное, при прогрессировании которого развивается острая дыхательная недостаточность. Лица с синдромом плохо слышат и видят, у них двоится в глазах и шумит в ушах. Со стороны органов зрения пациенты отмечают наличие светобоязни и боль при движении глазными яблоками. Офтальмологи часто обнаруживают анизокорию, спазм аккомодации или скотомы. Одним из основных симптомов синдрома является гипестезия – снижение чувствительности кожи лица и конечностей. Подобные патологическое изменения связаны с приглушенным реагированием рецепторов кожи на внешние раздражители: тепло или холод, уколы, удары. В тяжелых случаях нервное окончания вообще перестают воспринимать различные экзогенные воздействия.
- Сирингомиелический синдром — сложный симптомокомплекс, проявляющийся парестезией или онемением конечностей; изменением тонуса мышц и их гипотрофией, приводящей к миастеническим расстройствам; поражением периферических нервов, проявляющимся болью в конечностях; дисфункцией органов таза в виде затрудненной дефекации или самопроизвольного мочеиспускания; возможны артропатии — поражения суставов.
- У больных с пирамидальной недостаточностью снижается сила в нижних конечностях и способность к тонким движениям, ограничивается объем движений, повышается мышечный тонус — так называемая спастичность, например, спастическая походка. Повышение сухожильных рефлексов сочетается с одновременным снижением кожных рефлексов — брюшных. Возможно появление патологических рефлексов. У пациентов страдает мелкая моторика рук.
Любое неосторожное движение усиливает симптомы патологии, делает их более выраженными и яркими. Изменение положения головы — частая причина потери сознания.
Синдром Киари II типа имеет сходные клинические проявления. У новорожденных возникает паралич гортани, врожденный стридор, ночное апноэ, дисфагия, срыгивания, нистагм, гипертонус мышц рук, цианоз кожи. Аномалии III и IV типов не совместимы с жизнью.
Диагностические мероприятия
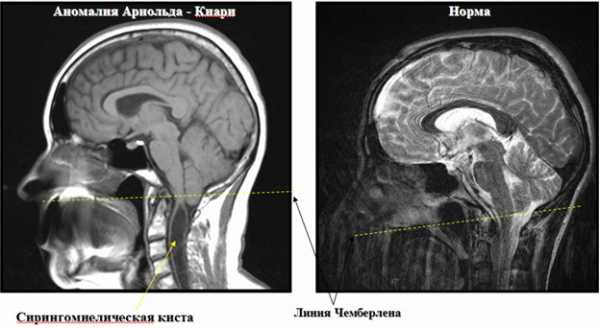
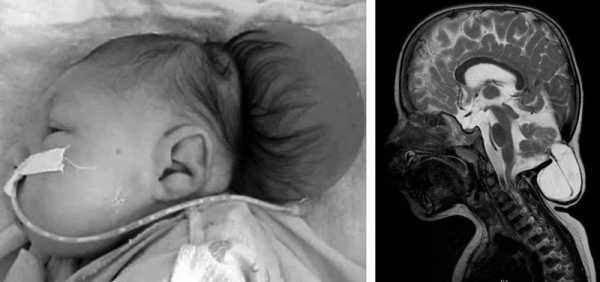
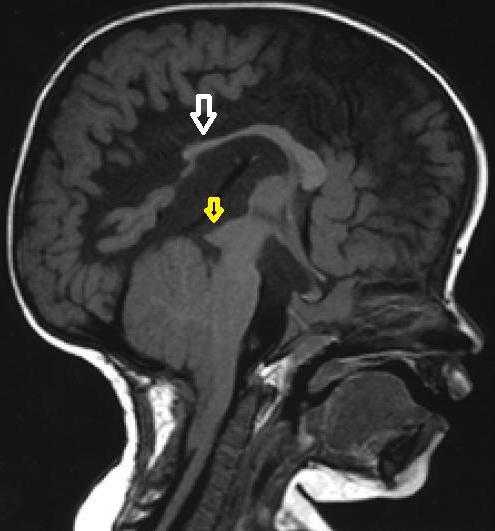
Аномалия Арнольда-Киари на снимке МРТ
Врачи-неврологи и невропатологи осматривают пациента и выявляют характерные особенности походки, изменение рефлексов и чувствительности на определенных участках тела, слабость в руках и прочие признаки. Все проявления мозжечкового, гидроцефального, бульбарного и прочих синдромов в совокупности позволяют врачу заподозрить аномалию.
После определения неврологического статуса больного требуется проведение комплексного неврологического обследования, включающего инструментальные методы — электроэнцефалографию, УЗИ головного мозга, реоэнцефалографию, ангиографию, рентгенографию. Эти методики выявляют лишь косвенные признаки патологии — изменения, происходящие в организме больного.
Ядерный магнитный резонанс лежит в основе особого нерентгенологического метода исследования – томографии. Этот спектроскопический анализ безопасен для большинства людей. Он дает изображение, состоящее из тонких срезов от магнитнорезонансного сигнала, проходящего через тело человека. На сегодняшний день именно МРТ позволяет быстро и точно поставить диагноз. Томография визуализирует структуру костей и мягкие ткани черепа, определяет пороки мозга и его сосудов.
Лечебный процесс
Лечение аномалии Киари комплексное, включающее медикаментозное воздействие, физиотерапевтические процедуры и хирургическое вмешательство. Именно оно в большинстве случаев помогает справиться с недугом и восстановить нормальную работу всего организма. Возможно применение средств народной медицины, которые дополняют, но не заменяют основное лечение. Использование фитосборов, отваров и настоев лекарственных трав должно быть одобрено лечащим врачом.
Лекарственная терапия и физиотерапия
Если больные испытывают сильную головную боль, боль в шее, мышцах и суставах, им назначают следующие группы препаратов:
- Обезболивающие средства – «Кеторол», «Пенталгин», «Анальгин».
- НПВС для уменьшения боли – «Мелоксикам», «Ибупрофен», «Вольтарен».
- Миорелаксанты для снятия напряжения с мышц шеи – «Мидокалм», «Сирдалуд».
Патогенетическое лечение синдрома включает:
- Препараты, улучшающие мозговое кровообращение – «Пирацетам», «Винпоцетин», «Циннаризин».
- Диуретики для уменьшения образования ликвора и с целью дегидратации – «Фуросемид», «Маннитол».
- Витамины группы В, поддерживающие работу нервной системы на оптимальном уровне и оказывающие антиоксидантное действие – «Тиамин», «Пиридоксин». Наиболее распространенные витаминные средства «Мильгамма», «Нейромультивит», «Комбилипен».
Если состояние больного признают крайне тяжелым, то его госпитализируют сразу в реанимационное отделение. Там пациента подключают к аппарату ИВЛ, устраняют имеющийся отек мозга, предупреждают инфекционные патологии и корректируют неврологические нарушения.
Физиотерапевтическое воздействие дополняет медикаментозное лечение, позволяет быстрее добиться положительных результатов, ускоряет процессы восстановления функций организма и выздоровления больных. Неврологи назначают:
- Криотерапию, оказывающую обезболивающий эффект, стимулирующую работоспособность желез внутренней секреции и укрепляющую иммунитет.
- Лечение лазером, улучшающее трофику и микроциркуляцию в очаге поражения.
- Магнитотерапию, оказывающую общее оздоравливающее действие и запускающую внутренние резервы организма.
В настоящее время особой популярностью пользуется кинезиологическая терапия, которая направлена на развитие умственных способностей и достижение физического здоровья через двигательные упражнения. Ее также включают в схему лечения данного синдрома.
Лечение не проводится вообще, если патология была обнаружена случайно, во время прохождения томографического обследования совсем по другому поводу, и у больного отсутствуют какие-либо характерные симптомы. За состоянием таких пациентов специалисты ведут динамическое наблюдение.
Оперативное вмешательство
Стойкие неврологические нарушения с парестезиями, дистонией мышц, параличами и парезами требуют проведения хирургической коррекции. Оперативное вмешательство показано также в тех случаях, когда медикаментозная терапия на дает положительного результата. Операции преследуют одну цель — устранение сдавления и ущемления мозга, а также восстановление нормальной циркуляции ликвора.
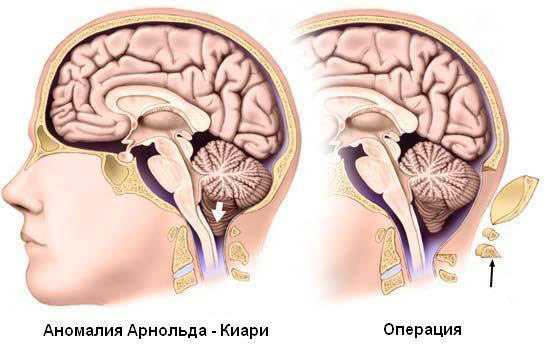
В настоящее время нейрохирурги спасают жизнь больным путем выполнения декомпрессивных и шунтирующих операций. В первом случае выпиливают часть затылочной кости с целью расширения большого отверстия, а во втором создают обходной путь для оттока ликвора по имплантационным трубкам с целью снижения его объема и нормализации внутричерепного давления.
После хирургического вмешательства всем пациентам показаны реабилитационные мероприятия. Если лечение было успешным, у больных восстанавливаются утраченные функции — дыхательные, двигательные, сердечно-сосудистые, нервные. В течение трех лет возможно рецидивирование патологии. В таких случаях больных признают инвалидами.
Видео: об операции при синдроме Арнольда-Киари
Народная медицина
Народные средства, применяемые при данной патологии, устраняют боль и расслабляют напряженные мышцы. Они эффективно дополняют традиционную терапию синдрома.
Наиболее популярные средства:
- Настой алтея для постановки компрессов,
- Прогревания пораженного места горячим куриным яйцом,
- Медовые компрессы,
- отвар папоротника или малины для приема внутрь.
Синдром Арнольда-Киари – порок развития, протекающий в бессимптомной форме или проявляющийся клинически с момента рождения. Патология имеет весьма разнообразную симптоматику и подтверждается с помощью МРТ. Лечебный подход к каждому пациенту индивидуален. Тактика лечения варьируется от симптоматического воздействия медикаментами до оперативного вмешательства с трепанацией черепа и удалением части мозговых структур.
Профилактика
Поскольку этиология синдрома окончательно не выяснена и нет конкретной информации о его патогенезе, предупредить развитие патологии не представляется возможным. Будущим родителям необходимо знать все о ведении здорового образа жизни и при планировании беременности стараться соблюдать указанные правила:
- Отказаться от пагубных привычек в виде табакокурения и употребления спиртных напитков,
- Обогащать свой рацион белковыми продуктами, фруктами, овощами, ягодами, исключив из него сладости и вредности,
- Своевременно обращаться к врачам за медицинской помощью,
- Принимать лекарственных препараты по назначению врача и в строго указанных дозировках,
- С профилактической целью принимать поливитамины,
- Беречь свое здоровье и наслаждаться жизнью.
Прогноз патологии неоднозначный. Консервативное лечение часто не дает положительных результатов. Хирургическое вмешательство, выполненное своевременно и в полном объеме, не всегда восстанавливает утраченные функции организма. Согласно статистическим данным эффективность такого лечения редко превышает 50-60%. Синдром третьей степени имеет неблагоприятный прогноз, поскольку поражаются многие мозговые структуры. При этом возникают несовместимые с жизнью функциональные нарушения.
Видео: лекция по синдрому Арнольда-Киали
Аномалия Арнольда Киари 1 типа
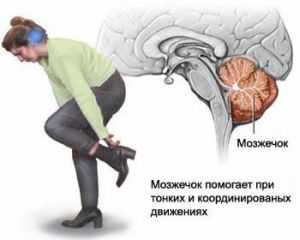
Не секрет, что во время формирования эмбриона в материнском лоне у него могут возникнуть разные патологии, в том числе и аномалия Арнольда Киари 1 типа. Что это за болезнь? Медики называют это явление отклонением, которое развивается у плода из-за несоответствия размеров части черепной коробки и мозга, размещённого в ней. Речь идёт о зоне, где расположен мозжечок. В результате происходит смещение в верхний отдел позвоночника части миндалин с дальнейшим их ущемлением.
Причины возникновения
Сможет ли нормально существовать человек, если у него обнаружена аномалия Арнольда Киари 1 типа? Продолжительность жизни, утверждают медики, зависит от нескольких факторов: степени тяжести и формы заболевания. Перед тем как ставить окончательный диагноз, они определяют причины, которые привели к развитию отклонения:
- Врождённые. Ещё в утробе у плода происходит деформация костей черепа: ямка, в которой находится мозжечок, оказывается слишком мала для растущего и развивающегося серого вещества. Среди других врождённых причин называют сильное увеличение затылочного отверстия у плода во время формирования скелета.
- Приобретённые. В первую очередь это травмы головы во время родов. Также к этой категории относят повреждения позвоночника: негативное влияние на него ликвора – особой жидкости, возникновение водянки и других патологий спинного мозга.
 Если быть предельно откровенными, точные причины учёные пока не могут назвать. Единственное, в чём они уверены: синдром не связан с хромосомными аномалиями развития.
Если быть предельно откровенными, точные причины учёные пока не могут назвать. Единственное, в чём они уверены: синдром не связан с хромосомными аномалиями развития.Факторы риска
Всем известно, что во время первого триместра беременности у плода идёт закладка фактически всех органов. Поэтому правильный образ жизни матери очень важен для нормального развития ребёнка. Если женщина не соблюдает элементарных правил, в дальнейшем это вызывает множество осложнений. Что касается синдрома Киари, то риск его возникновения увеличивают следующие факторы:
- Вредные привычки мамы: курение, злоупотребление алкоголем и наркотиками.
- Употребление сильнодействующих лекарств, самолечение.
- Перенесённые вирусные инфекции, особенно краснуха.
 При особо лёгких формах болезни рождённый малыш не чувствует до конца жизни особых симптомов, особенно если у него диагностирована аномалия Арнольда Киари 1 типа. Сколько живут дети, у которых третья или даже четвёртая стадия заболевания? Врачи дают неутешительные прогнозы. К сожалению, такие малыши в большинстве случаев обречены на летальный исход. Даже если у младенца обнаружен первый или второй тип недуга, ему необходимо лечение: эффективность хирургического вмешательства составляет 85 %.
При особо лёгких формах болезни рождённый малыш не чувствует до конца жизни особых симптомов, особенно если у него диагностирована аномалия Арнольда Киари 1 типа. Сколько живут дети, у которых третья или даже четвёртая стадия заболевания? Врачи дают неутешительные прогнозы. К сожалению, такие малыши в большинстве случаев обречены на летальный исход. Даже если у младенца обнаружен первый или второй тип недуга, ему необходимо лечение: эффективность хирургического вмешательства составляет 85 %.Типы синдрома
Независимо от своей тяжести, все они связаны с очень низким расположением мозжечка. Чем глубже и больше миндалины опускаются в позвоночный канал, тем более опасна и непредсказуема болезнь: вместе с ней диагностируют и другие отклонения. На основе этого и выделяют 4 формы патологии:
- Аномалия Арнольда Киари 1 типа - что это за разновидность болезни? Обычно она не вызывает какие-либо другие проблемы в развитии малыша. Структурных изменений в головном мозге не происходит, миндалины расположены в шейном отделе.
- Синдром второго типа. Мозжечок частично смещён в затылочное отверстие, при этом у эмбриона наблюдаются различные аномалии позвоночника, головного и спинного мозга.
- Заболевание третьего типа. Структуры заднего мозга полностью смещаются в затылочное отверстие. В этой зоне образуется грыжа.
- Заболевание Арнольда Киари четвертого типа. Для него характерна гипоплазия мозжечка – его недоразвитие. При этом он не смещается и часто сочетается с гидроцефалией и врождёнными кистами задней черепной ямы.
Медики говорят, что отклонения 2-го, 3-го и 4-го типа часто диагностируют в комбинации с другими серьёзными отклонениями в развитии малыша: гетеротопией коры мозга, гипоплазией подкорковых структур, аномалиями мозолистого тела и так далее.
Основные симптомы
Как уже говорилось, если стадия болезни очень лёгкая, а головной мозг задет незначительно, то болезнь может протекать фактически всю жизнь, не беспокоя человека. Но это достаточно редкий случай. Обычно перманентные и регулярные головные боли – главный признак такого недуга, как аномалия Арнольда Киари 1 типа. Симптомы для этой формы кроме мигрени такие: боль в шейном отделе, шум в ушах, тошнота и рвота, слабость рук, утрата чувствительности конечностей, мушки и двоение в глазах, неуверенная походка, смазанная речь и затруднённое дыхание.
Что касается синдрома второго типа, который является более опасным и сложным, то его характерные признаки проявляются сразу после рождения или в очень юном возрасте (часто в дошкольном). Обычно это свистящее шумное дыхание у младенцев, слабый крик, нарушение дыхания и глотания. Таких новорождённых срочно реанимируют и пытаются спасти путём хирургического вмешательства. В этих случаях главное - не упустить тот момент, когда ещё можно спасти жизнь ребёнку.
Другие признаки
Они наблюдаются при более тяжёлой степени синдрома. Первоисточниками инфаркта головного или спинного мозга часто бывают эти признаки аномалии Арнольда Киари: 1 тип не такой страшный, как его другие подвиды. Для них характерно не только двоение в глазах, но и полная слепота, потеря сознания, нарушение координации, тремор конечностей, проблемы с мочеиспусканием, слабость мышц, потеря чувствительности большой части тела (иногда целой половины туловища).

Развитие разных последствий для здоровья может спровоцировать аномалия Арнольда Киари 1 типа. Продолжительность жизни и её нормальное течение зависят от быстрой и точной диагностики. Единственным методом обнаружить патологию является МРТ (магнитно-резонансная томография) мозга. Она требует от пациента состояния полной неподвижности, поэтому активных младенцев усыпляют специальными медикаментами. При помощи этого аппарата также исследуют позвоночник, спинной мозг, шейные и грудные отделы скелета. МРТ позволяет обнаружить не только синдром, но и все патологии, которые сопутствуют болезни.
Осложнения
Развитие различных патологий провоцирует аномалия Арнольда Киари 1 типа. Последствия могут быть самыми разными: начиная от хронического арахноидита и заканчивая паренхиматозным поражением аксона. Особо ярко эти осложнения проявляются на фоне расстройства кровообращения в нервных тканях. Нейропсихические расстройства в этом случае проявляются поздно и проходят в форме отставания в развитии, параплегии. Если аномалия развивается очень прогрессивно, она приводит к гидроцефалии – чрезмерному накоплению жидкости в желудочковой системе мозга.
 Кроме того, синдром Арнольда Киари может вызвать такое сильное сдавливание спинного мозга, что человека полностью парализует. Под его негативным воздействием часто начинают образовываться кисты и полости в позвоночнике: в них поступает жидкость, что нарушает работу спинного мозга. Также возможны осложнения с дыханием вплоть до полной его остановки. Иногда медики регистрируют у пациентов застойную пневмонию – она становится последствием того, что человек теряет способность свободно двигаться.
Кроме того, синдром Арнольда Киари может вызвать такое сильное сдавливание спинного мозга, что человека полностью парализует. Под его негативным воздействием часто начинают образовываться кисты и полости в позвоночнике: в них поступает жидкость, что нарушает работу спинного мозга. Также возможны осложнения с дыханием вплоть до полной его остановки. Иногда медики регистрируют у пациентов застойную пневмонию – она становится последствием того, что человек теряет способность свободно двигаться.Консервативное лечение
Синдром лечат двумя методами. Врачи выбирают консервативный или хирургический путь в зависимости от симптомов и признаков, проявляющихся у пациента, степени тяжести состояния человека, осложнений и последствий болезни. Иногда исключительно при помощи медикаментов можно бороться с таким заболеванием, как аномалия Арнольда Киари 1 типа. Что это за терапия? Во-первых, медик постоянно проводит профилактические осмотры больного, направляет его на консультации к разным специалистам, которые после соответствующего консилиума принимают необходимые меры.
Если пациент жалуется на боли в затылочной области головы или в шейном отделе, ему назначают обезболивающие и противовоспалительные средства, а также препараты, которые помогают расслабить мускулатуру. Очень полезна и специальная лечебная физкультура – комплекс упражнений, направленный на устранение дрожи в конечностях и нормализацию координации. В этих целях также прописывают активную трудотерапию. А ещё будут полезны занятия с логопедом, который сумеет устранить проблемы с речью. Все эти методы могут отсрочить или полностью предотвратить проведение более агрессивного лечения – операции.
Хирургическое вмешательство
Случается, что вышеназванные меры не помогают – активно продолжает развиваться и доставлять кучу хлопот аномалия Арнольда Киари 1 типа. Лечение тогда кардинально меняется: врач начинает готовить человека к оперативному вмешательству. Нейрохирургическая операция проводится с тремя целями. Во-первых, для устранения таких серьёзных признаков болезни, как потеря сознания, расстройство зрения, слабость мускулатуры. Во-вторых, вмешательство помогает убрать первоисточник симптомов – ликвидировать сдавливание головного мозга. В-третьих, при помощи операции приводят в норму движение ликвора.
Вмешательство приостанавливает процесс изменений в структуре позвоночника и серого вещества: в результате симптомы ослабевают, пациент начинает чувствовать себя намного лучше. Обычно хирурги из задней части черепной ямы удаляют фрагмент кости: пространство для мозга увеличивается, серое вещество перестаёт смещаться и сдавливаться. Также падает внутричерепное давление. Кроме того, врачи активно используют такие методы, как шунтирование и закрытие позвоночного канала.
Профилактика
Чтобы у ребёнка не было вышеназванных проблем, будущая мама должна серьёзно относиться к своему положению. Во-первых, она обязана ознакомиться со всеми возможными патологиями, которые могут возникнуть у плода: нарушения развития нервной трубки, синдром Дауна, аномалия Арнольда Киари 1 типа. Что это за болезни, почему они возникают и какие меры можно принять для уменьшения риска развития этих и других отклонений, должна знать каждая будущая мать. Грамотная и осведомлённая беременная женщина будет максимально избегать всех тех факторов, которые негативно влияют на формирование эмбриона.

Во-вторых, женщина обязана полноценно питаться, употреблять разнообразные полезные блюда: рыбу, мясо, фрукты, овощи, крупы и молочные продукты. Категорически не допускается алкоголь. Кроме того, нужно отказаться от курения, не говоря уже о наркотиках и сильнодействующих препаратах. Любые лекарства, даже гомеопатические, будущая мама принимает только после назначения врача. В-третьих, женщина также должна много гулять, дышать свежим воздухом, много двигаться. Про нервы, переживания и негативные эмоции стоит забыть, наслаждаясь своим положением и получая максимальную радость от жизни.
Синдром Арнольда-Киари. Что такое аномалия Арнольда-Киари 1 типа
Закрыть- Болезни
- Инфекционные и паразитарные болезни
- Новообразования
- Болезни крови и кроветворных органов
- Болезни эндокринной системы
- Психические расстройства
- Болезни нервной системы
- Болезни глаза
- Болезни уха
- Болезни системы кровообращения
- Болезни органов дыхания
- Болезни органов пищеварения
- Болезни кожи
- Болезни костно-мышечной системы
- Болезни мочеполовой системы
- Беременность и роды
- Болезни плода и новорожденного
- Врожденные аномалии (пороки развития)
- Травмы и отравления
- Симптомы
- Системы кровообращения и дыхания
- Система пищеварения и брюшная полость
- Кожа и подкожная клетчатка
- Нервная и костно-мышечная системы
- Мочевая система
- Восприятие и поведение
- Речь и голос
- Общие симптомы и признаки
- Отклонения от нормы
- Диеты
- Снижение веса
- Лечебные
- Быстрые
- Для красоты и здоровья
- Разгрузочные дни
- От профессионалов
- Монодиеты
- Звездные
- На кашах
- Овощные
- Детокс-диеты
- Фруктовые
- Модные
- Для мужчин
- Набор веса
- Вегетарианство
- Национальные
- Лекарства
- Антибиотики
- Антисептики
- Биологически активные добавки
- Витамины
- Гинекологические
- Гормональные
- Дерматологические
- Диабетические
- Для глаз
- Для крови
- Для нервной системы
- Для печени
- Для повышения потенции
- Для полости рта
- Для похудения
- Для суставов
- Для ушей
- Желудочно-кишечные
- Кардиологические
- Контрацептивы
- Мочегонные
- Обезболивающие
- От аллергии
- От кашля
- От насморка
- Повышение иммунитета
- Противовирусные
- Противогрибковые
- Противомикробные
- Противоопухолевые
- Противопаразитарные
- Противопростудные
- Сердечно-сосудистые
- Урологические
- Другие лекарства
- Врачи
- Клиники
- Справочник
- Аллергология
- Анализы и диагностика
- Беременность
- Витамины
- Вредные привычки
- Геронтология (Старение)
- Дерматология
- Дети
- Женское здоровье
- Инфекция
- Контрацепция
- Косметология
- Народная медицина
- Обзоры заболеваний
- Обзоры лекарств
- Ортопедия и травматология
- Питание
- Пластическая хирургия
- Процедуры и операции
- Психология
- Роды и послеродовый период
- Сексология
- Стоматология
- Травы и продукты
- Трихология
- Другие статьи
- Словарь терминов
- [А] Абазия .. Ацидоз
- [Б] Базофилы .. Богатая тромбоцитами плазма
- [В] Вазектомия .. Выкидыш
- [Г] Галлюциногены .. Грязи лечебные
- [Д] Дарсонвализация .. Дофамин
- [Ж] Железы .. Жиры
- [З] Заместительная гормональная терапия
- [И] Игольный тест .. Искусственная кома
- [К] Каверна .. Кумарин
- [Л] Лапароскоп .. Лучевая терапия
- [М] Магнитотерапия .. Мутация
- [Н] Наркоз .. Нистагм
- [О] Общий анализ крови .. Отек
- [П] Паллиативная помощь .. Пульс
- [Р] Реабилитация .. Родинка (невус)
- [С] Секретин .. Сыворотка крови
- [Т] Таламус .. Тучные клетки
- [У] Урсоловая кислота
- [Ф] Фагоциты .. Фитотерапия
- [Х] Химиотерапия .. Хоспис
- [Ц] Цветовой показатель крови .. Цианоз
- [Ш] Штамм
- [Э]
что это такое, как проявляется, диагностика и лечение патологии

Врачи выделяют три вида аномалии Киари. Они зависят от изменений в структуре тканей мозга, проникающей в позвоночный отдел, а также от вида нарушений в работе и функционировании мозга спинного и головного.
Обычно симптомов этого заболевания нет, так что и лечить его не надо. Часто этот синдром обнаруживается только при исследовании других болезней.
Несмотря на кажущуюся безобидность, последствия аномалии Арнольда Киари могут быть плачевными.
Что это за явление
Мальформация Арнольда Киари встречается редко. При этом заболевании задняя часть мозга перемещается в каудальное направление, выпадает в затылочное отверстие.
В этом месте человек может ощущать тянущую боль, иногда могут возникать неврологические отклонения. Решение этой проблемы одно – операция, обычно выполняют шунтирование или декомпрессию ямки.
Классификация аномалии

Аномалия Арнольда Киари иметь разную тяжесть, поэтому есть четыре вида аномалии.
- Для первого типа характерно опущение миндалин мозжечка, они находятся ниже отверстия в затылке. Обнаружить это можно у подростка или взрослого человека. При такой стадии может быть выявлена гидромиелия, когда в центральном канале спинного мозга оказывается жидкость.
- Для второго типа характерно проявление сразу же с рождения ребенка. Ее можно заподозрить, если через затылок выходят миндалины мозжечка и продолговатый мозг. При таком заболевании всегда есть жидкость в спинном мозге, а иногда еще может быть врожденная спинномозговая грыжа.
- Для третьего типа характерно особое положение мозжечка и продолговатого мозга в менингоцеле шейно-затылочного отдела.
- У четвертого типа диагностируется почти полное отсутствие мозжечка. Причем он не опускается. Это состояние еще называют синдромом Денди-Уокера, когда к этой патологии прибавляется гидроцефалия, сирингомиелия, иногда могут появляться кисты на задней части черепа.
При втором и третьем типе аномалии Арнольда Киари могут быть другие симптомы со стороны нервной системы:
- полимикрогирия;
- гетеротопия мозга;
- кисты в отверстии Можанди;
- изменение мозолистого тела;
- изменений работы подкорки;
- изменение строения сильвиевого водопровода;
- может появиться намет и серп мозжечка.
Симптоматика
Типичными признаками аномалии Арнольда Киари являются:
- Частые боли в районе затылка, которая становится сильнее в моменты кашля и чихания.
- Боли в голове из-за повышения давления или напряжения шеи.
- Головокружения, потеря сознания, если резко повернуть голову или встать.
- Падение зрения.
- Плохое самочувствие.
- Температура тела ниже нормы, боли в руках.
- Слабость в мышцах рук.
- Спастичность пальцев.
- Наблюдается апноэ.
- Становится трудно глотать.
- Мозг перестает контролировать движения зрачков.
- Проблемы с мочеиспусканием.
- Подергивания глаз.
- Шум в ушах, если наклониться резко или повернуться.
- Тремор рук и ног.
- Проблемы с координацией.
- Проблемы в развитии мелкой моторики.
- Потеря чувствительности тела.
- Слабеют мышцы, из-за чего становится сложно ходить или поднимать вещи.
- Инфаркт мозга (спинного или головного).
Причины появления

До сих пор нет точного ответа, откуда пришло это заболевание. Некоторые неврологи говорят, что дело в неправильном размере черепа человека, из-за того, что в ней мало места, мозг не вмещается, и его части выходят за пределы черепной коробки.
Также есть вариант, что это явление вызвано слишком большим размером мозга, за которым не успевает расти голова, поэтому орган вынужден размещаться иным способом, перемещая мозжечок за пределы черепа.
Есть еще одна версия аномалии Арнольда Киари. При незначительной выраженности заболевания долгое время оно может резко обостриться из-за гидроцефалии. Жидкость влияет на размер желудочков мозжечка, увеличивая размер этого отдела мозга.
Мальформация не дает нормально развиваться связочному аппарату затылочно-шейной части тела. Поэтому при ударе или другой травме мозг страдает еще сильнее, появляются симптоматика манифестации.
Если во время беременности у женщины была эктопия шейки матки, то есть большая вероятность того, что у ребенка будет обнаружен этот синдром. Если обнаружить это на ранних сроках, можно прервать беременность. Другие причины пока не выявлены.
Осложнения
У людей с синдромом Арнольда Киари может долго не наблюдаться серьезных патологий, но если заболевание начнет развиваться, это может привести с тяжелым последствиям.
Среди них:
- скопление жидкости в черепной коробке вокруг мозга;
- развитие паралича, если жидкость соберется вокруг спинного мозга, причем не всегда в этой ситуации помогает операция;
- сирингомиелия – киста в позвоночнике, где есть жидкость. Это образование давит на спинной мозг, нарушая его работу;
- пороки сердца наблюдаются редко;
- проблемы с дыханием;
- пневмония из-за застоя;
- низкий уровень интеллекта.
Диагностика
 Для постановки диагноза надо знать степень развития заболевания, чтобы скорректировать тактику лечения. Также важен очный осмотр и несколько процедур: эхо- и электро- ЭГ мозга, рэоэнцефалография.
Для постановки диагноза надо знать степень развития заболевания, чтобы скорректировать тактику лечения. Также важен очный осмотр и несколько процедур: эхо- и электро- ЭГ мозга, рэоэнцефалография.
Но их результаты не могут досконально подтвердить диагноз, потому что они показывают только уровень внутричерепного давления.
Рентген черепа нужен, чтобы выявить аномалии в строении костей, которые характерны для этого заболевания.
КТ и МСКТ мозга не дают возможность врачам хорошо рассмотреть участок краниовертебрального перехода, а также не показывают состояние мягкого вещества мозга в задней части черепа.
Поэтому для диагностики аномалии Арнольда Киари используется магнитно-резонансная томография, с помощью которой можно увидеть состояние и головного, и спинного мозга.
Если надо обследовать ребенка, ему введут успокоительное, потому что в процессе процедуры нужна полная неподвижность.
Кроме МРТ мозга, может понадобиться исследование области шеи и грудной клетки. Это бывает в том случае, если есть вероятность обнаружения кист на позвоночнике, давящих на спинной мозг.
МРТ является лучшим выбором и потому, что может показать незначительные отклонения в работе нервной системы, которые укажут на присутствие болезни.
Лечение
Использование медикаментов при аномалии Арнольда Киари оправдано только в том случае, если пациент жалуется только на боль в затылке или шее. Разработаны препараты, которые снимают воспаление и обезболивают.
Если же это не помогает, а болезнь прогрессирует, придется соглашаться на операцию. Хирурги устранят все возможные проявления аномалии, которые давят на мозг, могут восстановить циркуляцию ликвора.
Обычно есть два варианта операций:
- рассечение концевой нити;
- декомпрессия отверстия в затылке, или краниоктомия.
Рассечение концевой нити
Плюсы такой тактики лечения аномалии Арнольда Киари операции такие:
- Работа хирурга длится всего 45 минут. Все действия не требуют серьезного инвазивного вмешательства, при этом можно вернуть на место все части мозга.
- После операции восстановление происходит быстро, реабилитация не нужна.
- После процедуры исключается смерть человека от этой аномалии.
- Устраняются не только последствия, но и причины появления мальформации, а также ряда других заболеваний.
- Смертность от такой процедуры нулевая.
- После операции не начнется гидроцефалиия из-за смещения миндалины.
- Человек чувствует себя лучше, а болезнь не развивается.
- Начинает лучше работать кровообращение, нервная система восстанавливает свои функции.
- Возможность жить полной жизнью.
Есть и некоторые минусы у этой операции:
- Небольшой шрам на копчике.
- После операции место разреза будет болеть несколько дней.
- Из-за ухода спастичности кажется, что стало меньше силы в конечностях.
- Из-за улучшения кровоснабжения мозга может увеличиться активность этого органа.
- Во время восстановления человек может чувствовать себя некомфортно, но это состояние быстро проходит.
Краниотомия
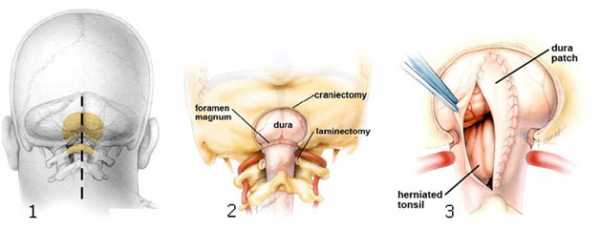
Схема оперативного лечения аномалии Арнольда Киари
Плюсы такой операции по декомпрессии затылка:
- Результат виден спустя пару дней после процедуры.
- Точно не наступит быстрая смерть от этого заболевания.
Но недостатков у декомпрессии при аномалии Арнольда Киари намного больше:
- Причина аномалии не исчезает.
- Смертность может достигать трех процентов.
- Работа хирурга оказывает сильное воздействие на организм, пациент может стать инвалидом.
- Улучшение есть, но не такое большое, как при первом методе лечения.
- Может развиться отек мозга.
- Возможно появление пневмоэнцефалии.
- После операции возможно появление гидроцефалии.
- Тетрапарез рук и ног.
- Во время процедуры может уйти порядка 14% от объема спинномозговой жидкости.
- Эмболия.
- Неврологический дефицит в редких случаях, зависит от зоны вмешательства.
- Появление инфекции в черепной коробке, из-за которой начнется менингит или появится абсцесс в мозге.
- Гемодинамические изменения ствола мозга.
- Появление эпидуральной гематомы.
- Кровоизлияние в мозге.
- Интрааксиальное кровоизлияние, из-за которого начинается неврологический дефицит.
Перед любым видом операции врачи тщательно обследуют пациента, чтобы понять, какой вариант будет более актуален в данном случае, а также изучают особенности организма, которые могут стать противопоказанием для хирургии.
Люди с такой аномалией могут жить долго, но не наслаждаться жизнью сполна. Поэтому операции показаны, чтобы дать возможность прочувствовать все прелести жизни без ограничений.
У всех методов лечения есть свои показания, поэтому всегда выбирается тот метод, который меньше всего навредит человеку.
Аномалия арнольда киари 1,2,3 степени
 Аномалия Арнольда-Киари – это нарушение функций мозга. Оно проявляется с самого рождения и идентифицируется отличием от нормальных параметров затылочной черепной ямки и составляющих мозга, которые расположены в этом районе.
Аномалия Арнольда-Киари – это нарушение функций мозга. Оно проявляется с самого рождения и идентифицируется отличием от нормальных параметров затылочной черепной ямки и составляющих мозга, которые расположены в этом районе.
Такие нарушения ведут к тому, что миндалины мозжечка опускаются в затылочную часть и ущемляются там.
В этом случае они спускаются таким образом, что достигают уровня первого или второго шейных позвонков и блокируют нормальное протекание спинно-мозговой жидкости.
На данный момент причины возникновения этого заболевания полностью не изучены, хотя сейчас их выделяют несколько разновидностей.
Аномалия Арнольда Киари 1 степени – это врождённые изменения, которые передались от родителей. Аномалия Арнольда Киари 2 степени – это травмы, которые получил ребёнок при рождении.
Статистика говорит, что от этого заболевания страдают в 3,3-8,2 случаев из 100000 наблюдавшихся. В среднем чаще от этого заболевания страдают люди 25-40 лет.
Типы болезни и особенности каждого из них
В 1891 году учёным Киари были выделены четыре вида данного заболевания. Эта классификация актуальна и в наши дни, поэтому врачи ею активно пользуются:
- Аномалия Арнольда Киари 1 типа— для него характерно снижение вдоль оси позвоночного столба составляющих затылочной черепной ямки до уровня первого или второго позвонка.
- II тип – очень часто можно встретить развитие гидроцефалии.
- III тип – встречается не так часто, как первые два, и характеризуется значительным перемещением большинства составляющих задней черепной ямки.
- IV тип – происходит гипоплазия мозжечка, при этом он не смещается вниз.
Если у человека наблюдается аномалия III или IV типа, то жить с ней невозможно.
Причины возникновения болезни
Нельзя наверняка сказать из-за чего может развиться аномалия Арнольда-Киари. Может быть, что вероятность рождения ребёнка с этим заболеванием увеличивают такие факторы, которые имеют влияние на организм женщины, которая ждёт ребёнка:
- употребления лекарственных препаратов выше допустимой нормы;
- размеры задней черепной ямки слишком малы;
- приём алкогольных напитков и курение в период беременности;
- частое поражение организма вирусными простудными заболеваниями;
- возникновение повреждений черепного отдела или головного мозга как результат родовой травмы;
- бывают врождённые и приобретённые причины появления аномалии Арнольда-Киари.
Симптомы и признаки
Чаще всего можно встретить аномалии Арнольда-Киари I типа. Зачастую она даёт о себе знать в период полового созревания или уже у взрослых. В таком случае проявляются следующие симптомы аномалии Арнольда-Киари:
- головные боли, возникающие в результате кашля, чихания или физических нагрузок;
- нарушение нормальной походки, вызванное проблемами с поддержанием равновесного состояния;
- проблемы с мелкой моторикой рук;
- нарушенная температурная чувствительность;
- онемение в области кистей рук;
- ощущение болей в области затылка и шеи.
Что касается II и III типов этого заболевания, то они проявляются такими же симптомами, только это происходит ещё с самого рождения.
В чём состоит сложность диагностики?
Очень сложно диагностировать аномалию Арнольда-Киари. Если делать неврологическое обследование, то оно сможет указать только на повышение уровня внутричерепного давления, которое называется гидроцефалией.
Если делать рентген головного мозга, то он диагностирует только нарушение строения костей, которыми обычно сопровождается это заболевание.
Единственный метод, который даёт возможность точно определить аномалию Арнольда-Киари – это магнитно-резонансная томмография. Для того, чтобы получить правильный и достоверный результат, нужно чтобы пациент лежал неподвижно. Для этого дети должны находится в медикаментозном сне.
Иногда это заболевание может себя не проявлять абсолютно никаким образом. И тогда его можно диагностировать только в результате усиленного медицинского осмотра.
На фото МРТ при аномалии Арнольда Киари
Лечение болезни
Существует два способа лечения аномалии Арнольда-Киари – консервативный или нехирургический и хирургический.
В случае, когда это заболевание протекает без проявления каких-либо симптомов, тогда оно не нуждается в лечении.
Если аномалия Арнольда-Киари проявляется только болевыми ощущениями в затылочной области и шее, то в этом случае назначается консервативное лечение. Тогда назначаются обезболивающие или противовоспалительные препараты. Это и есть нехирургический способ лечения.
В случае, когда симптомы болезни не поддаются консервативному лечению, тогда назначают хирургическое.
Целью такого лечения является устранение факторов, из-за которых происходит сжимание структур головного мозга.
В результате проведения операции расширяется затылочная часть головы, улучшается передвижение цереброспинальной жидкости по организму. Возможно проведение шунтирующих операций.
Осторожно, видео операции! Кликните, чтобы открыть
Осложнения, которые может вызвать заболевание
В некоторых случаях аномалия Арнольда-Киари может быть очень прогрессивно развивающимся заболеванием и привести к определенным осложнениям. Ими могут быть:
- Гидроцефалия, возникающая в результате накопления большего чем допускается количества жидкости.
- Паралич, причиной которого может выступать сдавливание спинного мозга.
- Сирингомиелия, что подразумевает образование полости или кисты в позвоночнике. В такие полости, после их возникновения, может поступать жидкость и приводить к нарушению нормальной работы спинного мозга.
- Возможно нарушение дыхательного процесса, которое развивается вплоть до его остановки.
- Могут быть случаи застойной пневмонии, возникающие как результат того, что пациент теряет способность передвигаться самостоятельно.
Прогноз
Бывают случаи, когда аномалия Арнольда-Киари I типа не проявляет свои симптомы на протяжении всей жизни человека, имеющего это заболевание.
Аномалия Арнольда-Киари III типа чаще всего приводит к неблагоприятному исходу.
Если при аномалии Арнольда-Киари I и III типов возникают какие-либо новрологические симптомы, то необходимо как можно раньше провести хирургическое лечение.
Это нужно потому, что такой неврологический недостаток очень сложно полностью устранить даже после успешного проведения операция, особенно если она была несвоевременной или затянутой.
Статистика показывает, что эффективность проведения хирургического лечения составляет 50-85%.
Аномалия Киари - причины, симптомы, диагностика и лечение
Аномалия Киари (синдром Арнольда-Киари) — заболевание, при котором структуры головного мозга, расположенные в задней черепной ямке, опущены в каудальном направлении и выходят через большое затылочное отверстие. В зависимости от типа аномалия Киари может проявляться головной болью в затылке, болью в шейном отделе, головокружением, нистагмом, обмороками, дизартрией, мозжечковой атаксией, парезом гортани, снижением слуха и ушным шумом, нарушением зрения, дисфагией, дыхательными апноэ, стридором, расстройствами чувствительности, гипотрофией мышц и тетрапарезом. Аномалия Киари диагностируется путем проведения МРТ головного мозга, шейного и грудного отделов позвоночника. Аномалия Киари, сопровождающаяся стойким болевым синдромом или неврологическим дефицитом, подлежит хирургическому лечению (декомпрессия задней черепной ямки или шунтирующие операции).
Общие сведения
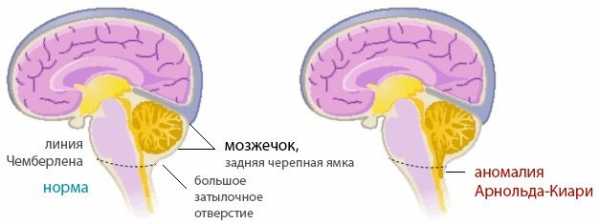
В области соединения черепа с позвоночным столбом находится большое затылочное отверстие, на уровне которого ствол головного мозга переходит в спинной мозг. Выше этого отверстия локализуется задняя черепная ямка. В ней расположен мост, продолговатый мозг и мозжечок. Аномалия Киари связана с выходом части анатомических структур задней черепной ямки в просвет большого затылочного отверстия. При этом происходит сдавление находящихся в этой области структур продолговатого и спинного мозга, а также нарушение оттока цереброспинальной жидкости из головного мозга, приводящее к гидроцефалии. Вместе с платибазией, ассимиляцией атланта и др. аномалия Киари относится к врожденным порокам развития краниовертебрального перехода.
Аномалия Киари встречается по различным данным у 3-8 человек на 100 тысяч населения. В зависимости от типа аномалия Киари может диагностироваться в первые дни после рождения ребенка или стать неожиданной находкой у взрослого пациента. В 80% случаев аномалия Киари сочетается с сирингомиелией.
Аномалия Киари
Причины возникновения аномалии Киари
До сих пор аномалия Киари остается заболеванием, об этиологии которого в неврологии нет единого мнения. Ряд авторов считает, что аномалия Киари связана с уменьшенным размером задней черепной ямки, приводящим к тому, что по мере роста расположенных в ней структур они начинают выходить через затылочное отверстие. Другие исследователи предполагают, что аномалия Киари развивается в результате увеличенных размеров головного мозга, который при этом как бы выталкивает содержимое задней черепной ямки через затылочное отверстие.
Спровоцировать переход незначительно выраженной аномалии в выраженную клиническую форму может гидроцефалия, при которой за счет увеличения желудочков увеличивается общий объем мозга. Поскольку аномалия Киари наряду с дисплазией костных структур краниовертебрального перехода сопровождается недоразвитием связочного аппарата этой области, любая черепно-мозговая травма может приводить к усугублению вклинения миндалин мозжечка в затылочное отверстие с манифестацией клинической картины заболевания.
Классификация аномалии Киари
Аномалия Киари подразделяется на 4 типа:
Аномалия Киари I характеризуется опущением миндалин мозжечка ниже большого затылочного отверстия. Обычно она проявляется у подростков или во взрослом возрасте. Зачастую сопровождается гидромиелией — скоплением цереброспинальной жидкости в центральном канале спинного мозга.
Аномалия Киари II проявляется в первые дни после рождения. Кроме миндалин мозжечка при этой патологии через большое затылочное отверстие выходят также червь мозжечка, продолговатый мозг и IV желудочек. Аномалия Киари II типа намного чаще сочетается с гидромиелией, чем первый тип, и в подавляющем большинстве случаев связана с миеломенингоцеле — врожденной спинномозговой грыжей.
Аномалия Киари III отличается тем, что опустившиеся через большое затылочное отверстие мозжечок и продолговатый мозг, располагаются в менингоцеле шейно-затылочной области.
Аномалия Киари IV заключается в гипоплазии (недоразвитии) мозжечка и не сопровождается его смещением в каудальном направлении. Некоторые авторы относят эту аномалию к синдрому Денди-Уокера, при котором гипоплазия мозжечка сочетается с наличием врожденных кист задней черепной ямки и гидроцефалией.
Аномалия Киари II и Киари III часто наблюдается в комбинации с другими дисплазиями нервной системы: гетеротопией коры головного мозга, полимикрогирией, аномалиями мозолистого тела, кистами отверстия Можанди, перегибом сильвиевого водопровода, гипоплазией подкорковых структур, намета и серпа мозжечка.
Симптомы аномалии Киари
Наиболее часто в клинической практике встречается аномалия Киари I типа. Она проявляется ликворногипертензионным, церебеллобульбарным и сирингомиелическим синдромами, а также поражением черепно-мозговых нервов. Обычно аномалия Киари I манифестирует в период полового созревания или уже во взрослом возрасте.
Для ликворногипертензионного синдрома, которым сопровождается аномалия Киари I, характерна головная боль в затылке и шейной области, усиливающаяся во время чихания, кашля, натуживания или напряжения мышц шеи. Может наблюдаться рвота, не зависящая от приема пищи и ее характера. При осмотре пациентов с аномалией Киари выявляется повышенный тонус мышц шеи. Среди мозжечковых нарушений наблюдаются нарушение речи (дизартрия), нистагм, мозжечковая атаксия.
Поражение ствола мозга, расположенных в нем ядер черепно-мозговых нервов и их корешков проявляются снижением остроты зрения, диплопией, расстройством глотания, снижением слуха по типу кохлеарного неврита, системным головокружением с иллюзией вращения окружающих предметов, ушным шумом, синдромом сонных апноэ, повторяющимися кратковременными потерями сознания, ортостатическим коллапсом. Пациенты, у которых имеется аномалия Киари, отмечают усиление головокружения и ушного шума при поворотах головой. Поворот головы у таких больных может спровоцировать обморок. Может отмечаться атрофические изменения половины языка и парез гортани, сопровождающийся осиплостью голоса и затруднением дыхания. Возможен тетрапарез с большим снижением мышечной силы в верхних конечностях, чем в нижних.
В случаях, когда аномалия Киари I сочетается с сирингомиелией, наблюдается сирингомиелический синдром: нарушения чувствительности по диссоциированному типу, онемения, мышечные гипотрофии, тазовые нарушения, нейроартропатии, исчезновение брюшных рефлексов. При этом некоторые авторы указывают на несоответствие размера и местонахождения сирингомиелической кисты распространенности расстройств чувствительности, степени выраженности парезов и мышечной гипотрофии.
Аномалия Киари II и Киари III имеют сходные клинические проявления, которые становятся заметны с первых минут жизни ребенка. Аномалия Киари II сопровождается шумным дыханием (врожденный стридор), периодами кратковременной остановки дыхания, двусторонним нейропатическим парезом гортани, нарушением глотания с забросом жидкой пищи в нос. У новорожденных аномалия Киари II проявляется также нистагмом, повышением мышечного тонуса в верхних конечностях, цианозом кожных покровов, возникающим во время кормления. Двигательные расстройства могут быть выражены в различной степени и прогрессировать вплоть до тетраплегии. Аномалия Киари III имеет более тяжелое течение и зачастую является не совместимым с жизнью нарушением развития плода.
Диагностика аномалии Киари
Неврологический осмотр и стандартный перечень неврологических обследований (ЭЭГ, Эхо-ЭГ, РЭГ) не дают специфических данных, позволяющих установить диагноз «аномалия Киари». Как правило, они выявляют лишь признаки значительного повышения внутричерепного давления, т. е. гидроцефалию. Рентгенография черепа выявляет только костные аномалии, которыми может сопровождаться аномалия Киари. Поэтому до внедрения в неврологическую практику томографических методов исследования диагностика этого заболевания представляла для невролога большие затруднения. Теперь врачи имеют возможность поставить таким пациентам точный диагноз.
Следует отметить, что МСКТ и КТ головного мозга при хорошей визуализации костных структур краниовертебрального перехода не позволяют достаточно точно судить о мягкотканных образованиях задней черепной ямки. Поэтому единственным достоверным методом диагностики аномалии Киари на сегодняшний день является магнитно-резонансная томография. Ее проведение требует обездвиженности пациента, поэтому у маленьких детей она проводится в состоянии медикаментозного сна. Кроме МРТ головного мозга для выявления менингоцеле и сирингомиелических кист необходимо также проведение МРТ позвоночника, особенно его шейного и грудного отделов. При этом проведение МРТ исследований должно быть направлено не только на диагностику аномалии Киари, но и на поиск других аномалий развития нервной системы, которые часто с ней сочетаются.
Лечение аномалии Киари
Бессимптомно протекающая аномалия Киари не нуждается в лечении. В случаях, когда аномалия Киари проявляется лишь наличием болей в шее и затылочной области, проводят консервативную терапию, включающую анальгетические, противовоспалительные и миорелаксирующие препараты. Если аномалия Киари сопровождается неврологическими нарушениями (парезы, расстройства чувствительности и мышечного тонуса, нарушения со стороны черепно-мозговых нервов и пр.) или не поддающимся консервативной терапии болевым синдромом, то показано ее хирургическое лечение.
Наиболее часто в лечении аномалии Киари применяется краниовертебральная декомпрессия. Операция включает расширение затылочного отверстия за счет удаления части затылочной кости; ликвидацию сдавления ствола и спинного мозга за счет резекции миндалин мозжечка и задних половин двух первых шейных позвонков; нормализацию циркуляции цереброспинальной жидкости путем подшивания в твердую мозговую оболочку заплаты из искусственных материалов или аллотрансплантата. В некоторых случаях аномалия Киари лечится при помощи шунтирующих операций, направленных на дренирование цереброспинальной жидкости из расширенного центрального канала спинного мозга. Цереброспинальная жидкость может отводиться в грудную или брюшную полость (люмбоперитонеальное дренирование).
Прогноз аномалии Киари
Важное прогностическое значение имеет тип, к которому относится аномалия Киари. В некоторых случаях аномалия Киари I может на протяжении всей жизни пациента сохранять бессимптомное течение. Аномалия Киари III в большинстве случаев приводит к летальному исходу. При появлении неврологических симптомов аномалии Киари I, а также при аномалии Киари II большое значение имеет своевременное проведение хирургического лечения, поскольку возникший неврологический дефицит плохо восстанавливается даже после успешно проведенной операции. По различным данным эффективность хирургической краниовертебральной декомпрессии составляет 50-85%.
причины, симптомы, типы и лечение
Педиатр Анна Колинько о патологии развития головного мозга, которая может встречаться у 30 % населения
Синдром хронической усталости, головокружения и боль в шее могут быть следствием мальформации (аномалии) Арнольда — Киари. После начала широкого использования МРТ стало понятно, что болезнь встречается у 14–30 % популяции
Мальформация Арнольда — Киари (МАК) — это патология развития ромбовидного мозга: продолговатого и заднего мозга, в последний входит Варолиев мост и мозжечок. При МАК задняя черепная ямка не соответствует мозговым структурам, расположенным в этой области: мозжечок и продолговатый мозг из‑за небольших размеров опускаются ниже большого затылочного отверстия, что приводит к их ущемлению и нарушению ликвородинамики. МАК относят к группе кранио-вертебральных (черепно-позвоночных) мальформаций.
В эпоху до МРТ частота МАК оценивалась от 3,3 до 8,2 наблюдений на 100 000 населения, а у новорожденных — 1 на 4–6 тысяч. Сегодня понятно, что распространенность синдрома Арнольда — Киари значительно больше. Из-за бессимптомного течения и в результате учета разных типов МАК цифры очень разнятся — от 14 до 30 %.
Все первые описания мальформации были посмертными. В 1883 году шотландский анатом Джон Клеланд (J. Cleland, 1835–1925 гг.) впервые описал удлинение ствола и опущение миндалин мозжечка в большое затылочное отверстие у 9 умерших новорожденных. В 1891 году австрийский патолог Ганс фон Киари (H. Chiari, 1851–1916 гг.) подробно охарактеризовал 3 типа мальформации у детей и взрослых. А в 1894 году немецкий патолог Юлиус Арнольд (J. Аrnold, 1835–1915 гг.) подробно описал синдром Киари 2 типа, в сочетании со спинномозговой грыжей (spina bifida). В 1896 году Киари дополнил свою классификацию четвертым типом. В 1907 году ученики Арнольда использовали термин «мальформация Арнольда — Киари» по отношению к аномалии 2 типа. Теперь это название распространилось на все типы. Некоторые врачи справедливо отмечают, что вклад Арнольда несколько преувеличен и верным будет термин «мальформация Киари».
Версии о причинах
Этиология и патогенез синдрома Арнольда — Киари остаются неуточненными. Киари предположил, что смещение мозжечка и продолговатого мозга происходит из‑за внутриэмбриональной гидроцефалии, которая возникает как следствие стеноза сильвиева водопровода — узкого канала длиной 2 см, который соединяет III и IV желудочки мозга.
Гидроцефалия — патологическое состояние, характеризующееся избыточным накоплением спинномозговой жидкости (ликвора) в ликворных пространствах головного мозга.
Клеланд полагал, что аномалия связана с первичным недоразвитием ствола головного мозга. В 1938 г. канадский нейрохирург Уайлдер Пенфилд (W. G. Penfield, 1891–1976 гг.) и его коллега предложили «теорию тяги»: в процессе роста фиксированный спинной мозг втягивает в полость позвоночного канала расположенные выше отделы. В «унифицированной» теории Дэвид Маклон (D. G. McLone) и Пол Неппер (P. A. Knepper) в 1989 году предположили, что первично возникает дефект нервной трубки с истечением ликвора и недостаточным расширением желудочковой системы, что приводит к формированию уменьшенной задней черепной ямки. Однако последующие исследования говорят о том, что существуют разные варианты патологии Арнольда — Киари: с уменьшением задней черепной ямки и без такового, с нарушением ликворооттока и без. Описаны семейные случаи МАК 2 типа, однако роль генетических факторов еще недостаточно изучена.
Типы мальформаций
-
1 тип — опущение миндалин мозжечка в позвоночный канал ниже уровня большого затылочного отверстия с отсутствием спинномозговой грыжи. У 15–20 % пациентов этот тип сочетается с гидроцефалией, а у 50 % больных — с сирингомиелией — заболеванием, при котором в спинном и продолговатом мозге образуются полости. В 1991 году было предложено подразделить аномалии Арнольда — Киари 1 типа на тип А — с сирингомиелией и тип В — без сирингомиелии.
Сирингомиелии при Арнольде — Киари 1 степени.
Энцефаломенингоцеле — врожденная грыжа головного мозга и его оболочек, содержащая цереброспинальную жидкость.
Спинальная дизрафия — порок развития, заключающийся в отсутствии слияния по средней линии парных закладок кожи, мускулатуры, позвонков, спинного мозга
-
2 тип — опущение нижних отделов червя мозжечка, продолговатого мозга и IV желудочка. Отличительным признаком данного типа является сочетание со спинномозговой грыжей (spina bifida) в поясничном отделе, отмечается прогрессирующая гидроцефалия, часто — стеноз водопровода мозга. Среди детей с менингомиелоцеле до 90 % случаев сопровождается аномалией Арнольда — Киари 2 степени.
Менингомиелоцеле — грыжа спинномозгового канала, при которой происходит выпячивание тканей и вещества спинного мозга через костный дефект позвоночного столба
- 3 тип — грубое смещение заднего мозга в позвоночный канал с высокой шейной или подзатылочной грыжей головного мозга и его оболочек и выраженным гипертензивно-гидроцефальным синдромом.
- 4 тип — гипоплазия (недоразвитие) мозжечка без смещения его вниз с эктопией продолговатого мозга.
- 0 тип. В 1998 году американский детский нейрохирург Берманс Искандер (В. J. Iskandar) с коллегами впервые ввел понятие «Киари 0» («Сhiari 0») в описании 5 пациентов, имеющих неврологические симптомы аномалии Арнольда — Киари с сирингомиелией и положением миндалин мозжечка на уровне большого затылочного отверстия. Этот тип так же называют «пограничным с Киари».
0, 1 и 2 степени синдрома Арнольда — Киари наиболее распространены в популяции. III и IV типы обычно несовместимы с жизнью.
Симптоматика
Неврологические симптомы 0 и 1 типов аномалии Арнольда-Киари наиболее часто начинают беспокоить в возрасте 20–40 лет. Степень дислокации миндалин мозжечка может нарастать под влиянием неблагоприятных факторов. Чаще всего жалобами при МАК 0 типа являются головная боль, преимущественно шейно-затылочной локализации, а также боль в шее. Аномалия Арнольда — Киари 1 типа у взрослых чаще проявляется жалобами на нистагм, дизартрию, атаксию, интенционный тремор (тремор при произвольных движениях), головную боль, головокружение, нарушение чувствительности, парезы, нарушение функции тазовых органов, нарушения частоты и ритма пульса, ритма дыхания, лабильность артериального давления, симптомы поражения каудальной группы черепных нервов (IX, X, XI, XII пары) — нарушение чувствительности лица и бульбарные расстройства (расстройства глотания и речи).
Синдром Арнольда-Киари 2 степени впервые проявляется не у взрослых, а у новорожденных или в раннем детском возрасте. МАК 2 типа протекает более тяжело, дети с такой патологией уже рождаются с гидроцефальной формой черепа. Гидроцефалия препятствует нормальному развитию. Кроме того, такие дети страдают нарушениями дыхания, сердцебиения и глотания. Часто заболевание сопровождается судорожными припадками. У детей развивается нистагм, апноэ, стридор, парез голосовых связок, дисфагия с регургитацией, нарушение тонуса в конечностях. Выраженность неврологической симптоматики в первую очередь зависит от выраженности нарушений ликвородинамики, а не от степени эктопии миндалин мозжечка.
Терапия
Лечение аномалий Арнольда — Киари зависит от выраженности неврологической симптоматики. Консервативная терапия включает в себя нестероидные противовоспалительные препараты и миорелаксанты. Если в течение 2–3 месяцев консервативная терапия безрезультатна или у пациента имеется выраженный неврологический дефицит, показано оперативное вмешательство. В процессе операции устраняется сдавление нервных структур и нормализуется ликвороток путем увеличения объема (декомпрессии) задней черепной ямки и установки шунта. Оперативное лечение эффективно, по разным источникам, в 50–85 % случаев, в оставшихся случаях симптоматика регрессирует не полностью. Операцию рекомендуется выполнять до развития тяжелого неврологического дефицита, поскольку восстановление происходит лучше при минимальных изменениях неврологического статуса. Подобное оперативное лечение выполняется почти в каждом федеральном нейрохирургическом центре России и проводится в рамках высокотехнологичной медицинской помощи по системе ОМС.
Пациенты, имеющие мальформацию Арнольда-Киари 0 и 1 типа, могут даже не знать о наличии у себя этого заболевания в течение всей жизни. Вследствие пренатальной диагностики МАК II, III и IV типа дети с данной патологией рождаются всё реже, а современные технологии выхаживания позволяют значительно увеличить продолжительность жизни таких детей.
Источники
- Мальформация Арнольда — Киари. Пренатальные и клинические наблюдения Авраменко Т. В., Шевченко А. А., Гордиенко И. Ю. Вестник ВГМУ. –2014. — № 2. — С. 87–95.
- Богданов Э. И. Аномалия Арнольда — Киари: патогенез, клинические варианты, классификация, диагностика и лечение / Э. И. Богданов, М. Р. Ярмухаметова // Вертеброневрология. — 1998. — № 2–3. — C. 68–73.
- Мальформация Арнольда — Киари: классификация, этиопатогенез, клиника, диагностика: (обзор литературы) / Л. А. Дзяк [и др.] // Украинский нейрохирургический журнал. — 2001. — № 1. — С. 17–23.
- Егоров О. Е. Клиника и хирургическое лечение аномалии Киари 1 типа / О. Е. Егоров, Г. Ю. Евзиков // Неврологический журнал. — 1999. — № 5. — С. 28–31.
- Кантимирова Е. А., Шнайдер Н. А., Петрова М. М., Строцкая И. Г., Дутова Н. Е., Алексеева О. В., Шаповалова Е. А. Встречаемость аномалии Арнольда — Киари в практике невролога. Неврология, Нейропсихиатрия, Психосоматика. — 2015. № 4. — С. 18–22.
- Латышева В. Я., Олизарович М. В., Филюстин А. Е., Гурко Н. А. Клинико-томографические соотношения при синдроме Арнольда — Киари. Международный неврологический журнал. 2011; (7): 6–11.
- Юркина Е. А. Клинико-неврологические и нейровизуализационные сопоставления при аномалии краниовертебральной области у взрослых. Диссертация на соискание ученой степени кандидата медицинских наук Санкт-Петербург, 2016.
что это такое, прогноз выживаемости
Из статьи вы узнаете особенности аномалии Арнольда Киари, симптомах заболевания, диагностике, лечении и прогнозе патологии.
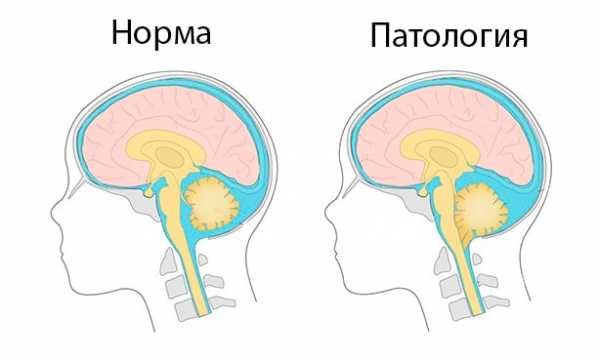
Аномалия Арнольда Киари – патологическое состояние, при котором мозговые структуры задней черепной ямки опускаются вниз через большое затылочное отверстие с симптомами сдавливания нейронов и нарушением оттока ликвора.
Общие данные
В области соединения черепа с позвоночным столбом находится большое затылочное отверстие, на уровне которого ствол головного мозга переходит в спинной мозг. Выше этого отверстия локализуется задняя черепная ямка. В ней расположен мост, продолговатый мозг и мозжечок. Аномалия Киари связана с выходом части анатомических структур задней черепной ямки в просвет большого затылочного отверстия. При этом происходит сдавление находящихся в этой области структур продолговатого и спинного мозга, а также нарушение оттока цереброспинальной жидкости из головного мозга, приводящее к гидроцефалии. Вместе с платибазией, ассимиляцией атланта и др. аномалия Киари относится к врожденным порокам развития краниовертебрального перехода.
Аномалия Киари встречается по различным данным у 3-8 человек на 100 тысяч населения. В зависимости от типа аномалия Киари может диагностироваться в первые дни после рождения ребенка или стать неожиданной находкой у взрослого пациента. В 80% случаев аномалия Киари сочетается с сирингомиелией.
Анатомические изменения при аномалии Арнольда-Киари
При данной патологии мозжечок находится в задней черепной ямке. В норме нижняя часть мозжечка (миндалины) должна располагаться выше большого затылочного отверстия. При аномалии Арнольда-Киари миндалины расположены в позвоночном канале, то есть под большим затылочным отверстием. Большое затылочное отверстие служит своеобразной границей между позвоночником и черепом, а также между спинным и головным мозгом. Над этим отверстием расположена задняя черепная ямка, а под ним – позвоночный канал. Нижний отдел ствола мозга (продолговатый мозг) переходит в мозг спинной на уровне большого затылочного отверстия.
В норме ликвор (спинномозговая жидкость) должен свободно циркулировать в субарахноидальных пространствах спинного и мозгового мозга. Эти субарахноидальные пространства соединяются между собой на уровне большого затылочного отверстия, что обеспечивает свободный отток спинномозговой жидкости от головного мозга. При аномалии Арнольда-Киари миндалины расположены под большим затылочным отверстием, что затрудняет свободный ток ликвора между головным и спинным мозгом. Миндалины мозжечка блокируют большое затылочное отверстие подобно пробке, что существенно нарушает отток спинномозговой жидкости и ведет к развитию гидроцефалии.
Типы аномалии
В 1891 году Киари определил четыре основных типа патологии и подробно описал каждую. Данной классификацией врачи пользуются и по сегодняшний день.
- 1 тип. Для него характерно опущение структур задней черепной ямки ниже плоскости большого затылочного отверстия.
- 2 тип, для которого характерна каудальная дислокация нижних отделов черепа, 4-го желудочка и продолговатого мозга. Нередко это сопровождается гидроцефалией.
- 3 тип. Данный тип заболевания является довольно редким и для него характерно грубое каудальное смещение всех структур задней черепной ямки.
- 4 тип, когда гипоплазия мозжечка происходит без его смещения вниз.
Третий и четвертый типы аномалии Арнольда-Киари лечению не поддаются и, как правило, приводят к летальному исходу. У преимущественного большинства пациентов (около 80%) аномалия Арнольда-Киари сочетается с сирингомиелией – патологией спинного мозга, для которой характерно образование кист в нем, способствующих развитию прогрессирующей миелопатии. Образуются подобные кисты при опущении структур задней черепной ямки и в результате сдавления шейного отдела спинного мозга.
Причины аномалии
Выделяют три анатомические причины порока:
- Размер черепной задней ямки меньше нормы.
- Головной мозг либо мозжечок быстрее увеличивается, чем растет череп.
- Недостаточно развит фиксирующий аппарат краниовертебрального перехода (связки вверху хребта).
Во всех случаях мозговая ткань выжимается в затылочное отверстие, к 1 шейному позвонку.
Патологические причины аномалии Арнольда-Киари:
- краснуха, прочие опасные инфекции во время беременности;
- прием лекарств с тератогенным и эмбриотоксическим действием;
- родовая травма черепа;
- осложнение родов — применение вакуума, щипцов, отказ от кесарева при узком тазе;
- черепно-мозговая травма — ДТП, нападение, иные несчастные случаи;
- интракраниальные или внутричерепные опухоли;
- водянка, иные болезни мозга, вызывающие смещение структур вниз;
- генетические расстройства.
Справка! Среди причин синдрома Арнольда-Киари называют алкогольную/наркотическую зависимость будущих отца и/или матери, которая появилась еще до зачатия ребенка.
Во время беременности к провоцирующим факторам аномалии ребенка относят вредные привычки матери, пассивное курение — пребывание в прокуренных местах, рядом с курящими. Вызвать нарушение развития может влияние радиации, канцерогены, действие токсических веществ.
Симптоматика, признаки патологии
Болезнь сопровождается специфическими синдромами. Выраженность и количество этих симптомокомплексов зависит от тяжести порока, какие внутричерепные и позвоночные структуры затронуты патологией.
Характеристика неврологических синдромов:
| Название синдрома | Характер проявления | Симптомы аномалии |
|---|---|---|
| Церебральный (мозжечковый) | Расстройство координации движений, неврологические нарушения, неконтролируемый нистагм (непроизвольное подергивание, колебание глаз) | Нет точности мелкой моторики, походка шаткая, неустойчивость вертикального положения тела. Тремор (дрожание) пальцев, ухудшение речи, двоение картинки (нарушение зрения). У детей отмечается умственная недостаточность |
| Гипертензионно-гидроцефальный | Нарушение оттока и всасывания ликвора, повышенное внутричерепное давление (интракраниальная гипертензия), водянка мозга | Распирающая головная боль. Тошнота, рвота вне зависимости от приема лекарств, пищи, напряжение мышц на затылке. Ухудшение самочувствия после пробуждения, движения головой. |
| Бульбарно-пирамидный | Сдавливание пирамид, моста, продолговатого мозга | Паралич либо парез (слабость) мышечной группы на руке, туловище. Боль/дискомфорт в зоне затылка. Усиливается во время чихания, движений головы. Онемение участков кожи, тела. Нарушение глотательной, дыхательной, речевой функции. |
| Вертебробазилярной недостаточности | Сдавливание кровеносных сосудов, ухудшение питания мозга, кислородное голодание тканей (гипоксия) | Приступы головокружения, полуобморочное состояние, потери сознания. Ухудшение мышечной силы. Нарушения зрения, слуха. |
| Корешковый | Поражение подъязычного, блуждающего, других нервов и ядер в зоне затылочной кости, большого отверстия, атланто-аксиального сустава, мозжечка | Онемение в области лица, языка. Нарушения глотания, речи, слуха. |
| Сирингомиелитический | Развитие кисты в веществе продолговатого/спинного мозга, сохраняется восприятие тела в пространстве | Пропадает на коже предплечий, рук, туловища чувствительность к раздражителям, смене температуры (нет боли от ожогов, ран, обморожения). Слабость и атрофия мышц конечностей, туловища, головы, деформация мелких суставов, искривление позвоночника. Возможно недержание кала, мочи, иная дисфункция выделительных органов. |
Справка! Аномалии Арнольда-Киари характерно как бессимптомное течение, так и медленное развитие, бурное прогрессирование. Признаки могут проявляться с рождения либо отсутствовать всю жизнь.
Признаки 1 типа аномалии
В случае симптомного течения болезни у человека наблюдается до трех синдромов: вертебробазилярной недостаточности, гипертензионно-гидроцефальный и/или мозжечковый. При этой тяжести порока Арнольда-Киари возможны регулярные приступы мигрени, распирающая боль в зоне затылка. Ухудшается четкость движений, мелкой моторики.
Наблюдаются признаки скачков давления и гипоксии:
- головокружение;
- перед глазами мелькание «мушек»;
- шум в ушах;
- слабость.
Человек ощущает дискомфорт сзади у основания черепа, при поворотах/наклонах головы. В случае прогрессирования возможно присоединение других симптомокомплексов. Это проявление бульбарно-пирамидного, корешкового и/или сирингомиелического синдрома.
Признаки 2 степени
При втором типе порока врачи выявляют одновременно минимум три неврологических синдрома. Родившийся ребенок мало двигается, слабо кричит, ручки согнуты, мышцы напряжены. Есть дисфункция нервной системы, органов.
Пороку Арнольда-Киари 2 типа характерно:
- систематическая остановка дыхания;
- ослабление глоточных мышц;
- синюшность кожи.
Осложняется вторая степень частичным либо полным параличом. Утрата двигательных функций отмечается в пальцах, руке/ноге, в области головы, всего тела.
Признаки 3 степени
Третьей степени мальформации Арнольда-Киари характерно проявление 5―6 синдромов одновременно. Ребенок не кричит сразу после рождения. У пациентов часто отмечается потеря слуха, зрения, дисфункция выделительных органов. Нет четкой координации движения, по утрам возникает рвота, есть аномалии строения позвоночника, головы.
Признаки 4 типа
У людей с четвертой степенью тяжести порока преобладает мозжечковый и гипертензионно-гидроцефальный синдром. Дети беспокойны, слабо вскрикивают, тихо плачут. Если выживают, на обследованиях выявляют снижение интеллекта, отсутствие нормального физического развития. В целом, симптомы с прогнозом жизни третьей и четвертой степени тяжести похожи.
Диагностика
Диагностика порока базируется на: данных анамнеза. Доктор обязательно уточнит, как проходила беременность, родовой период, было ли влияние токсинов, медикаментов; полученной нейровизуализационной картинке. Выполняют компьютерную либо магнитно-резонансную томографию. Причём МРТ более информативна, потому что даёт возможность увидеть заднюю черепную ямку и головной мозг, находящийся там. Применение КТ без внутривенного усиления, то есть введения рентгенконтрастного йодсодержащего средства, бессмысленно, — она не обнаружит мальформацию. О пороке свидетельствует «выпадение» нижних частей мозжечка из полости черепной коробки и изменение положения других стволовых структур, наблюдаемое на томограмме.
Особенности терапии
Выбор метода лечения при аномалии Арнольда-Киари зависит от наличия симптомов заболевания.
Если порок был выявлен случайно (то есть не имеет клинических проявлений и не беспокоит больного) при проведении магнитно-резонансной томографии по поводу другого заболевания, то лечение не проводят вообще. За пациентом устанавливается динамическое наблюдение, чтобы не пропустить момент появления первых клинических симптомов сдавления мозга.
Если аномалия проявляет себя незначительно выраженным гипертензионно-гидроцефальным синдромом, то предпринимаются попытки консервативного лечения. Для этой цели используют:
- дегидратационные препараты (мочегонные). Они уменьшают количество ликвора, способствую уменьшению болевого синдрома;
- нестероидные противовоспалительные средства с целью уменьшения болевого синдрома;
- миорелаксанты при наличии напряжения мышц в шейной области и другие препараты.
Медикаментозная терапия болезни Арнольда-Киари:
| Группа лекарств | Действие | Цель применения |
|---|---|---|
| Нестероидные противовоспалительные средства | Предупреждают и лечат воспаление тканей | Устраняют болевой синдром |
| Ненаркотические анальгетики + спазмолитики, препараты нейромышечной блокады | Обезболивают | |
| Миорелаксанты | Снимают мышечное напряжение | |
| Мочегонные, прочие лекарства дегидратационной терапии | Выводят жидкость, снижают давление | Частично снимают симптомы, вызванные скоплением ликвора/крови в основании черепа |
| Нейропротекторы | Предупреждают повреждение, гибель нервных клеток | Убирают кислородное голодание тканей, улучшают регенерацию и функционирование ЦНС, мозга |
| Метаболики | Нормализуют обменные процессы | |
| Витамины группы В | Противосудорожные, восполняют дефицит веществ, улучшают регенерацию клеток | |
| Антиоксиданты | Подавляют окисление, образование радикалов | |
| Вазодилататоры | Сосудорасширяющие | |
| Ангиопротекторы | Укрепляют сосудистые стенки |
Если применения лекарственных препаратов оказывается достаточно, то на какой-то период на этом и останавливаются. Если же эффекта нет, или у больного появляются признаки других неврологических синдромов (мышечная слабость, утрата чувствительности, признаки нарушения функции черепно-мозговых нервов, периодические приступы потери сознания и так далее), то тогда прибегают к хирургическому лечению.
Оперативное лечение
Операция состоит в выполнении трепанации задней черепной ямки, удалении части затылочной кости, резекции опущенных в большое затылочное отверстие миндалин мозжечка, рассечении спаек субарахноидального пространства, мешающих циркуляции ликвора.
Иногда может понадобиться шунтирующая операция, целью которой является отведение избытка спинномозговой жидкости. «Лишняя жидкость» по специальной трубке (шунту) сбрасывается в грудную или брюшную полость.
Определение момента, когда возникает необходимость в хирургическом лечении, — весьма важная и ответственная задача. Длительно существующие изменения чувствительности, утрата мышечной силы, дефекты черепно-мозговых нервов могут не восстановиться полностью после оперативного вмешательства. Поэтому важно не упустить момент, когда действительно без операции не обойтись.
При II типе порока оперативное лечение показано практически в 100% случаев без предварительного консервативного лечения.
Прогноз
Важное прогностическое значение имеет тип, к которому относится аномалия Киари. В некоторых случаях аномалия Киари I может на протяжении всей жизни пациента сохранять бессимптомное течение.
Аномалия Киари III в большинстве случаев приводит к летальному исходу. При появлении неврологических симптомов аномалии Киари I, а также при аномалии Киари II большое значение имеет своевременное проведение хирургического лечения, поскольку возникший неврологический дефицит плохо восстанавливается даже после успешно проведенной операции. По различным данным эффективность хирургической краниовертебральной декомпрессии составляет 50-85%.
Источники: krasotaimedicina.ru, neboleem.net, doktor-ok.com, doctor-neurologist.ru, ustamivrachey.ru
Аделина Павлова