Синдром эдвардса что это такое
Синдром Эдвардса - причины, симптомы, диагностика и лечение
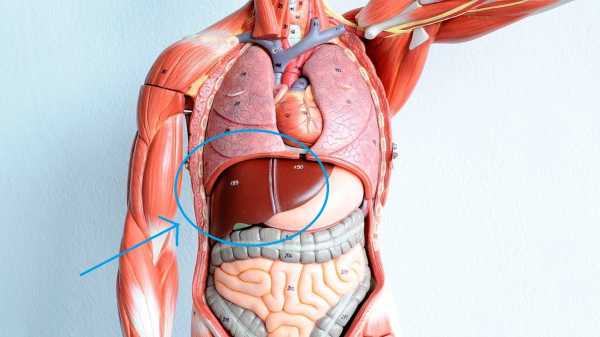
Синдром Эдвардса - хромосомное заболевание, обусловленное трисомией по 18-ой хромосоме и сопровождающееся множественными пороками развития. Для синдрома Эдвардса характерны своеобразные фенотипические признаки (долихоцефалическая форма черепа, микрофтальмия, недоразвитие ушных раковин, микроретрогнатия и др.), аномалии опорно-двигательной, сердечно-сосудистой, пищеварительной, мочеполовой системы, ЦНС. Синдром Эдвардса может быть диагностирован на этапе беременности (УЗИ-скрининг, инвазивная пренатальная диагностика) либо уже после рождения ребенка на основании внешних признаков и цитогенетического исследования. Дети с синдромом Эдвардса нуждаются в симптоматическом лечении и хорошем уходе.
Общие сведения
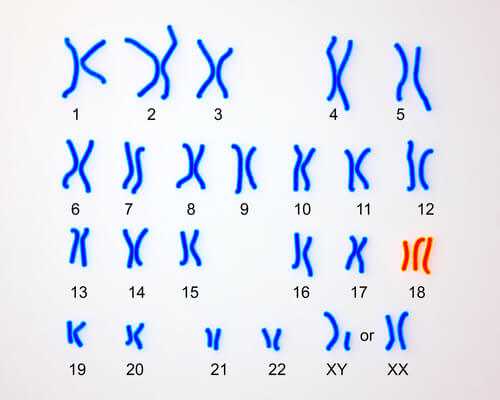
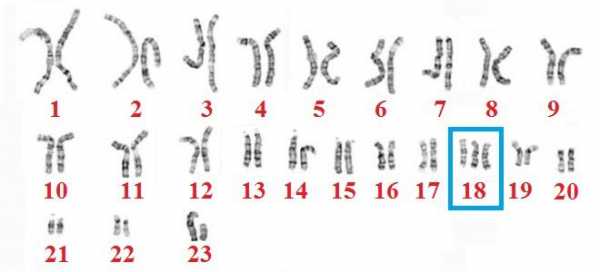
Синдром Эдвардса – количественная хромосомная аберрация, при которой имеет место частичная или полная трисомия по 18 аутосоме. Синдром получил название по имени генетика J. Edwards, подробно описавшего заболевание в 1960 г. и выделившего свыше 130 характерных для данной патологии симптоматических дефектов. Синдром Эдвардса – второе по распространенности хромосомное заболевание после синдрома Дауна; частота рождения детей с синдромом Эдвардса составляет 1:5000-7000. Примерно три четверти всех больных синдромом Эдвардса – девочки; предполагается, что большая часть беременностей плодом мужского пола заканчивается внутриутробной гибелью и самопроизвольным абортом.
Синдром Эдвардса
Причины синдрома Эдвардса
Развитие синдрома Эдвардса объясняется хромосомными нарушениями, происходящими на стадии гаметогенеза (овогенеза или сперматогенеза) либо дробления зиготы и приводящими к увеличению числа хромосом 18-й пары. В 80-90% случаев цитогенетические варианты синдрома Эдвардса представлены простой трисомией 18, реже - мозаичной формой или несбалансированными перестройками (транслокациями).
Причиной полной трисомии служит мейотическое нерасхождение хромосом. Практически во всех случаях лишняя хромосома является материнской по происхождению. Этот вариант синдрома Эдвардса является наиболее тяжелым по своим проявлениям и неблагоприятным в плане прогноза. Возникновение мозаицизма связано с нерасхождением хромосом на ранней стадии дробления зиготы. В этом случае лишнюю хромосому будут содержать не все клетки плода, а лишь их часть. Транслокация – присоединение части 18-ой хромосомы к другой паре может произойти как в процессе созревания гамет, так и после оплодотворения. При этом клетки организма содержат две гомологичные 18-е хромосомы и ее дополнительную часть, прикрепленную к другой хромосоме.
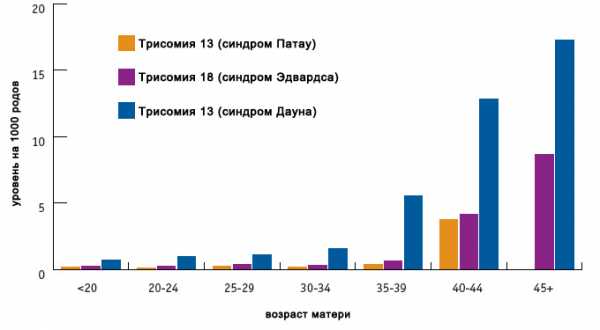
Как и в случае с синдромом Дауна, возраст матери является наиболее значимым риск-фактором рождения ребенка с синдромом Эдвардса. В редких случаях у родителей может выявляться носительство сбалансированной транслокации.
Симптомы синдрома Эдвардса
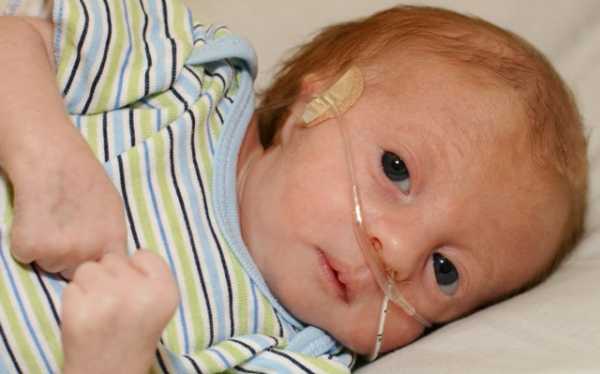
Во время беременности наблюдается многоводие, слабая активность плода, маленькая плацента, единственная пупочная артерия. Ребенок с синдромом Эдвардса рождается с низкой массой тела (около 2170 г) и пренатальной гипотрофией при доношенной или даже переношенной беременности. У части детей определяется состояние асфиксии при рождении.
У новорожденных с синдромом Эдвардса имеются характерные фенотипические признаки, позволяющие предположить данную хромосомную патологию. В первую очередь обращает на себя внимание долихоцефалическая форма черепа с преобладанием продольного размера над поперечным, низкий лоб, выступающий затылок, микрогнатия, маленький рот, микрофтальмия. У детей с синдромом Эдвардса часто встречаются расщелины верхней губы и нёба, эпикант, птоз, экзофтальм, косоглазие, короткая шея с избыточной кожной складкой. Типичные деформации ушных раковин включают маленькие мочки, отсутствие козелков, узкие слуховые проходы, низкое расположение ушей.
Внешний облик детей дополняется характерными для синдрома Эдвардса деформациями скелета - скрещенными пальцами кистей, укороченной грудиной, аномалиями ребер, врожденным вывихом бедра, косолапостью, «стопой-качалкой», синдактилией стоп и пр. У многих детей имеются гемангиомы и папилломы кожи.
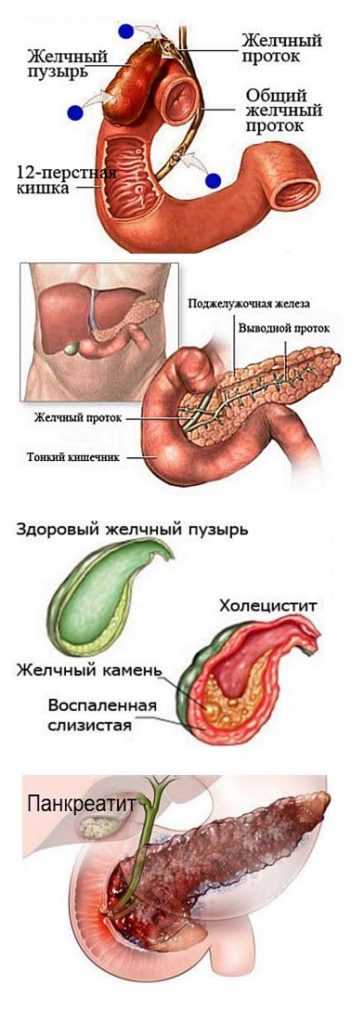
При синдроме Эдвардса имеются множественные тяжелые аномалии со стороны практически всех систем организма. Врожденные пороки сердца могут быть представлены дефектами межжелудочковой и межпредсердной перегородок, коарктацией аорты, транспозицией магистральных сосудов, дисплазией клапанов, тетрадой Фалло, аномальным дренажом легочных вен, декстракардией и др. При синдроме Эдвардса может выявляться патология развития желудочно-кишечного тракта: диафрагмальные, пупочные и паховые грыжи, дивертикул Меккеля, трахеопищеводные свищи, пилоростеноз, атрезия подвздошной кишки и ануса. Наиболее частыми аномалиями мочеполовой системы у детей с синдромом Эдвардса служат подковообразная почка, гидронефроз, дивертикулы мочевого пузыря, гипоспадия и крипторхизм (у мальчиков), двурогая матка, внутриматочная перегородка и гипертрофия клитора (у девочек).
Пороки развития центральной нервной системы характеризуются наличием микроцефалии, менингомиелоцеле, гидроцефалии, аномалии Арнольда-Киари, кист арахноидального сплетения, гипоплазии мозжечка и мозолистого тела. У всех выживших детей с синдромом Эдвардса имеются интеллектуальные нарушения - олигофрения в степени глубокой имбецильности или идиотии.
Новорожденные с синдромом Эдвардса испытывают трудности с сосанием, глотанием и дыханием, из-за чего им требуется зондовое питание или длительная ИВЛ. Дети с синдромом Эдвардса, как правило, погибают на первом году жизни из-за тяжелых врожденных пороков развития и связанных с ними осложнений (сердечно-сосудистой и дыхательной недостаточности, пневмонии, кишечной непроходимости и т. д.).
Диагностика синдрома Эдвардса
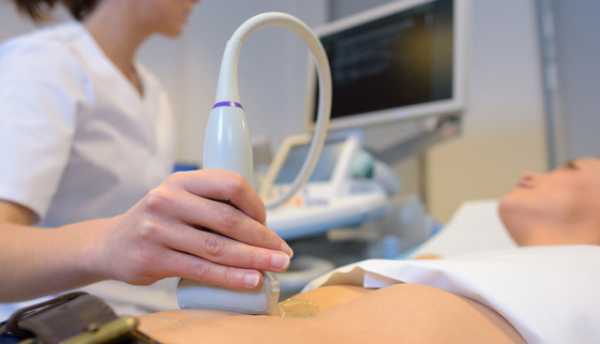
Важнейшей задачей диагностики служит антенатальное выявление синдрома Эдвардса у плода, поскольку данная патология является медицинским показанием для искусственного прерывания беременности. Заподозрить наличие синдрома Эдвардса можно в процессе УЗИ плода и допплерографии маточно-плацентарного кровотока по косвенным признакам (множественным аномалиям развития плода, агенезии пупочной артерии, малой величине плаценты, многоводию и пр.).
Наибольшую диагностическую значимость имеет стандартный пренатальный скрининг, включающий анализ крови на сывороточные маркеры: βХГЧ и PAPP на 11-13 неделе беременности; βХГЧ, альфа-фетопротеин и свободный эстриола на 20-24 неделе гестации.
При оценке степени риска рождения ребенка с синдромом Эдвардса учитываются данные биохимического и ультразвукового скрининга, срок беременности, возраст и масса тела женщины. Беременным, попадающим в группу высокого риска, предлагается проведение инвазивной дородовой диагностики (биопсии хориона, амниоцентеза, кордоцентеза) с последующим кариотипированием плода.
В случае рождения живого ребенка с синдромом Эдвардса необходимо как можно более раннее всестороннее обследование, направленное на выявление тяжелых пороков развития. Новорожденный с синдромом Эдвардса должен быть осмотрен неонатологом, детским кардиологом, детским неврологом, детским хирургом, детским ортопедом, детским урологом и др. Наиболее важными диагностическими исследованиями, которые должны быть выполнены ребенку с синдромом Эдвардса в первые часы жизни, служат эхокардиография, УЗИ органов брюшной полости и УЗИ почек.
Лечение синдрома Эдвардса
Поскольку в большинстве случаев аномалии развития оказываются несовместимыми с жизнью, лечение детей с синдромом Эдвардса сводится к оказанию симптоматической помощи, направленной на поддержание физиологических функций, продление жизни и улучшение ее качества. Хирургическая коррекция врожденных пороков, как правило, является рискованной и неоправданной.
Поскольку дети с синдромом Эдвардса ослаблены и подвержены частой заболеваемости инфекциями мочевыводящих путей, средним отитом, конъюнктивитом, синуситами, пневмониями и пр., они нуждаются в тщательно организованном уходе, полноценном питании, регулярном наблюдении со стороны педиатра.
Прогноз и профилактика синдрома Эдвардса
Во всех случаях прогноз при синдроме Эдвардса крайне неблагоприятный: в среднем мальчики живут 2-3 месяца, девочки – 10 месяцев. До 1 года доживает лишь 10% больных, до 10 лет – не более 1%. Относительно благоприятные шансы в отношении выживания имеют дети с мозаичной формой синдрома Эдвардса.
Риск рождения ребенка с синдром Эдвардса теоретически существует в любой супружеской паре; известно, что такая вероятность выше у возрастных родителей (для женщин старше 45 лет – 0,7%). С целью своевременного выявления хромосомной патологии у плода не следует пренебрегать антенатальным скринингом, входящим в программу введения беременности.
Синдром Эдвардса — Википедия
Материал из Википедии — свободной энциклопедии
Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 25 мая 2015; проверки требуют 16 правок. Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 25 мая 2015; проверки требуют 16 правок.Синдром Э́двардса (синдром трисомии 18) — хромосомное заболевание, характеризуется комплексом множественных пороков развития и трисомией 18 хромосомы. Описан в 1960 году Джоном Эдвардсом (John H. Edwards). Популяционная частота примерно 1:3000 в США, и 1:5000 в мире на 2016 год. Дети с трисомией в 18 хромосоме чаще рождаются у пожилых матерей, взаимосвязь с возрастом матери менее выражена, чем в случаях трисомии хромосомы 21[3] и 13[4]. Для женщин старше 45 лет риск родить больного ребёнка составляет 0,7 %. Девочки с синдромом Эдвардса рождаются в три раза чаще мальчиков. Выживание после года жизни составляет около 5–10%[5].
Причиной заболевания является наличие дополнительной 18-й хромосомы (трёх вместо двух в норме для диплоидного набора) в кариотипе зиготы.
Лишняя хромосома обычно появляется до оплодотворения. У человека нормальные половые клетки — гаметы — содержат по 23 хромосомы (гаплоидный набор) и, сливаясь, они дают кариотип зиготы — 46 хромосом. К появлению лишней хромосомы у гамет обычно приводит нерасхождение хромосом при мейотическом делении, вследствие чего в половой клетке оказывается 24 хромосомы. В случае, если такая клетка встретит при оплодотворении гамету от противоположного пола, они образуют зиготу с трисомией.
В одном случае из десяти наблюдается мозаицизм в явлении трисомии 18: лишнюю хромосому несут не все клетки организма. Это говорит о том, что нерасхождение произошло на ранней стадии развития зародыша, а все клетки с трисомией — потомки неправильно поделившейся клетки зародыша.
Дети с трисомией 18 рождаются с низким весом, в среднем около 2200 грамм, при этом длительность беременности — нормальная или даже превышает норму. Фенотипические проявления синдрома Эдвардса многообразны. Чаще всего возникают аномалии мозгового и лицевого черепа, мозговой череп имеет долихоцефалическую форму. Нижняя челюсть и ротовое отверстие маленькие. Глазные щели узкие и короткие. Ушные раковины деформированы и в подавляющем большинстве случаев расположены низко, несколько вытянуты в горизонтальной плоскости. Мочка, а часто и козелок отсутствуют. Наружный слуховой проход сужен, иногда отсутствует. Грудина короткая, из-за чего межреберные промежутки уменьшены и грудная клетка шире и короче нормальной. В 80 % случаев наблюдается аномальное развитие стопы: пятка резко выступает, свод провисает (стопа-качалка), большой палец утолщён и укорочен. Из дефектов внутренних органов наиболее часто отмечаются пороки сердца и крупных сосудов: дефект межжелудочковой перегородки, аплазии одной створки клапанов аорты и лёгочной артерии. У всех больных наблюдаются гипоплазия мозжечка и мозолистого тела, изменения структур олив, выраженная умственная отсталость, снижение мышечного тонуса, переходящее в повышение со спастикой.
Продолжительность жизни детей с синдромом Эдвардса невелика: 60 % детей умирают в возрасте до 3 месяцев, до года доживает лишь 5-10 %. Основной причиной смерти служат остановка дыхания и нарушения работы сердца. Оставшиеся в живых — глубокие олигофрены.
Частота появления синдрома Эдвардса составляет ~ 1:7000 зачатий и 1:8000 рождений живых детей. Риск рождения больного ребёнка увеличивается с возрастом, особенно, если мать болеет диабетом.
Кроме трисомии 18, присутствующей во всех клетках организма, а также мозаичной трисомии 18, возможна и частичная трисомия. При этом часть хромосомы 18 присоединяется к другой хромосоме. Такой эффект называется транслокация, и он может произойти как при созревании гамет, так и после оплодотворения в клетках зародыша. В клетках организма при этом оказываются две гомологичные хромосомы 18 и, дополнительно, часть хромосомы 18, прикреплённая к другой хромосоме. У людей, страдающих частичной трисомией 18, аномалии проявляются слабее, нежели при типичном синдроме Эдвардса.
- ↑ Disease Ontology release 2019-05-13 — 2019-05-13 — 2019.
- ↑ Monarch Disease Ontology release 2018-06-29sonu — 2018-06-29 — 2018.
- ↑ Синдром Дауна
- ↑ Синдром Патау
- ↑ Genetics Home Reference. Trisomy 18 (англ.). Genetics Home Reference. Дата обращения 20 сентября 2019.
Синдром Эдвардса: причины, признаки, диагностика, лечение
Синдром Эдвардса – заболевание, обусловленное спонтанной мутацией генов и появлением дополнительной 18 хромосомы. Подобные патологические изменения происходят в организме плода во время эмбриогенеза. Трисомия по 18-ой аутосоме сопровождается разнообразными пороками развития, приводящими к инвалидности или смерти ребенка. Синдром Эдвардса распространен по всему земному шару без четкой зависимости от местности или расы.
У больных детей изменяется внешний вид, поражается опорно-двигательная, пищеварительная, сердечно-сосудистая, нервная и мочеполовая системы. Следствием количественной хромосомной аберрации является своеобразный фенотип — длинная голова; недоразвитые уши, глаза и челюсти; короткая верхняя губа; косолапость. Синдром Эдвардса проявляется глубокой умственной отсталостью и многочисленными врожденными пороками внутренних органов: сердца, мозга, почек.
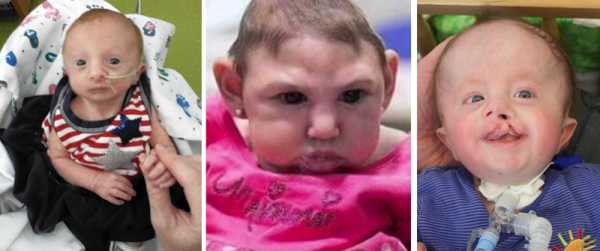
дети с синдромом Эдвардса
Впервые патологию описал ученый-генетик J. Edwards в 1960 году. Он выделил более 100 симптомов заболевания. Благодаря этому открытию синдром получил свое название. Синдром Эдвардса по распространенности среди хромосомных недугов уступает только синдрому Дауна. Трисомия – вариант хромосомной мутации, при которой у человека в клетках содержится не 46, а 47 хромосом. Существует всего 3 синдрома в данной группе нозологий: синдром Эдвардса (трисомия 18 хромосомы), Дауна (трисомия 21 хромосомы) и Патау (трисомия 13 хромосомы). При наличии других добавочных хромосом заболевание несовместимо с жизнью.
Согласно статистики, болеют синдромом Эдвардса преимущественно девочки. Женщины, носящие под сердцем плод мужского пола, обычно не вынашивают беременность – она заканчивается самопроизвольным абортом. Риск развития синдрома возрастает в случае беременности после 40 лет. Болезнь негативно сказывается на течении беременности: отмечается многоводие, недостаточная активность плода и поздние роды – после 42 недель.
Диагностика синдрома состоит из трех этапов — до зачатия, до родов и после рождения ребенка. Проводят ультразвуковое и цитогенетическое исследования плода, а также используют инвазивные методы, позволяющие выявить недуг пренатально. Новорожденных детей осматривают специалисты в области неонтологии, детской кардиологии, неврологии, хирургии, ортопедии, урологии. Им делают ЭКГ, УЗИ почек, исследование органов брюшной полости. Синдром Эдвардса — генетическое заболевание, вылечить которое невозможно. Больным детям требуется симптоматическая терапия и тщательный уход. Современные методы лечения способны поддержать жизнь ребенка на оптимальном уровне и добиться определенного прогресса в его развитии.
Большинство детей с синдромом Эдвардса погибают внутриутробно. Новорожденные девочки редко доживают до 8-10 месяцев, а мальчики — до 2-3- месяцев. Только 10% рожденных детей способны прожить год. Взрослыми становятся лишь единицы. Больные дети умирают от сердечной дисфункции, воспаления легких, удушья, кишечной непроходимости. Эти осложнения обусловлены врожденные пороками развития.
Синдром Эдвардса – показание к прерыванию беременности. Рождение больных деток является осознанным выбором родителей. Чаще всего так происходит у женщин, которые не становились на учет по беременности и не проходили рекомендуемых исследований. Обычно родители отказываются от больного ребенка, практически обреченного на гибель.
Причины
Кариотип здорового ребенка состоит из 46 хромосом: по 23 от обоих родителей. У лиц с синдромом Эдвардса под влиянием не установленных наукой факторов происходит дублирование генетического материала, и появляется дополнительная 47 хромосома, которая является «лишней». Обычно мутации подвергается 18 хромосома. Так формируется название – трисомия 18. Хромосомные нарушения происходят в процессе образования гамет или дробления зиготы. В большинстве случаев возникает простая трисомия 18, крайне редко – мозаичная или транслокационная форма. Причины синдрома Эдвардса в настоящее время остаются неизвестными. Больной ребенок может появиться на свет в семье, где родители и родственники являются абсолютно здоровыми.
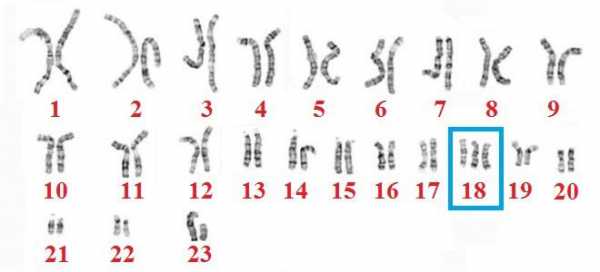
- Полная трисомия – три 18 хромосомы в каждой клетке плода. Патология обусловлена нерасхождением хромосом в процессе мейоза. Почти всегда лишняя хромосома передается по материнской линии. Этот вариант синдрома Эдвардса протекает довольно тяжело по сравнению с другими формами, встречается намного чаще и практически всегда имеет неблагоприятный прогноз.
- Мозаицизм связан с нерасхождением хромосом после слияния половых клеток на ранней стадии дробления зиготы. Обе гаметы изначально имеют нормальный набор хромосом, но в результате удвоения генетического материала и формирования зародыша происходит сбой. При этом только часть клеток плода получит лишнюю хромосому. Доля патологических клеток никогда не превышает 50%. Их число зависит от того, на каком этапе деления начальной клетки произошел сбой. Чем позже это происходит, тем меньше будет доля дефектных клеток. Общее состояние пациента при этом легче, чем при классической форме трисомии 18.
- Частичная трисомия или транслокация – добавление фрагмента третьей хромосомы в результате дефекта деления генетического материала. Транслокационная перестройка ведет к избыточности информации и нарушению генетической последовательности в двух хромосомах. Гены 18 хромосомы переходят с одного участка на другой. Для пациентов с частичной трисомией 18 прогноз лучше, чем для детей с полной формой, но все равно остается неблагоприятным.
Клинически все 3 варианта данного синдрома протекают по одному типу, но первый вариант все же может отличаться более тяжелой формой.
Факторы, способствующие развитию патологии:
- неблагоприятная экология,
- облучение,
- воздействие химикатов и прочих токсинов,
- прием алкоголя беременной женщиной,
- активное и пассивное курение,
- воздействие некоторых лекарств,
- кровное родство супругов,
- заболевания половой сферы,
- возраст матери старше 40 лет.
Вышеперечисленные факторы лишь повышают риск развития данной мутации, а не являются ее непосредственными причинами.
Передача измененного набора хромосом последующим поколениям невозможна. Большинство больных не доживают до репродуктивного возраста. Репродуктивные органы, как и репродуктивные способности у них недоразвиты. Синдром Эдвардса не передается по наследству.
Симптомы
Избыток генетической информации в клетках приводит к появлению соответствующих симптомов болезни, которые объединены под названием «синдром Эдвардса».
Первые признаки патологии появляются во время беременности:
- Многоводие,
- Слабая активность плода,
- Гипоплазия плаценты,
- Аномальное строение пуповины.
Новорожденные дети имеют низкую массу тела – около 2 кг, а также признаки гипотрофии при доношенной или переношенной беременности. У них нарушен процесс дыхания, сосания и проглатывания молока. Им требуется питание через зонд и длительная вентиляция легких.
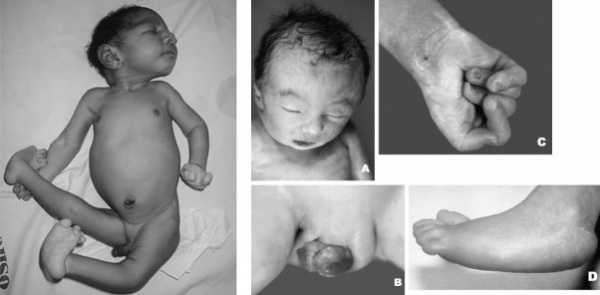
Фенотипические признаки новорожденных с синдромом Эдвардса:
- Долихоцефалия — длинноголовость,
- Непропорционально маленькая голова,
- Низкий лоб,
- Выступающий затылок,
- «Готическое» небо,
- Узкие глазные щели,
- Узкий и вдавленный нос,
- Микрогнатия — маленькая челюсть,
- Микрофтальмия — мелкие глазные яблоки,
- «Заячья губа» и «волчья пасть»,
- Вертикальная складка на внутреннем уголке глаза,
- Патологически неправильный прикус,
- Искаженные черты лица,
- Опущение верхнего века,
- Стробизм,
- Короткая шея,
- Деформация ушных раковин,
- Низкая посадка ушей.
Характерные деформации скелета:
- синдактилия кистей,
- укороченная и расширенная грудина,
- косолапость,
- искривление позвоночника,
- гипо- и атрофия мышц,
- «стопа-качалка»,
- полное слияние или перепончатость пальцев на нижних конечностях,
- аномальное сгибание и разгибание суставов,
- флексорное положение кисти.
Тяжелые аномалии со стороны внутренних органов:
- врожденные пороки сердца,
- грыжи,
- меккелев дивертикул,
- сужение привратника,
- отсутствие анального отверстия,
- удвоение мочеточника,
- почка в форме подковы,
- расширение чашечно-лоханочного комплекса,
- выпячивание стенки мочевого пузыря,
- неопущение яичка в мошонку,
- двурогая матка,
- клиторомегалия,
- атрофия или сглаживание извилин мозга,
- микроцефалия,
- менингомиелоцеле,
- водянка головного мозга,
- субарахноидальные кисты,
- недоразвитие мозжечка и мозолистого тела.
Нарушения психической сферы:
- умственная отсталость,
- олигофренизм,
- имбицилизм,
- идиотизм,
- задержка нервно-психического развития.
Тяжелейшие пороки фактически не дают больным шанса на выживание. Даже качественное лечение не спасает детей от гибели на первом году жизни.
Перечисленные клинические признаки синдрома Эдвардса довольно специфичны и разнообразны. Они позволяют заподозрить у пациента данный недуг и поставить предварительной диагноз. Сочетание наиболее частых симптомов с высокой вероятностью говорит о наличии у ребенка тяжелой патологии.
Диагностика
Поскольку синдром Эдвардса характеризуется довольно большим количеством ярко выраженных отклонений, его довольно просто диагностировать даже по внешним проявлениям. Однако этого недостаточно, чтобы поставить окончательный диагноз.
Диагностика синдрома Эдвардса складывается из трех этапов — обследование супружеских пар до момента зачатия, беременной женщины до родов и ребенка после появления на свет.
Диагностика до зачатия ребенка — идеальный вариант, но не всегда применимый. Специалисты-генетики могут лишь предположить, каков риск рождения ребенка с хромосомным заболеванием в данной семье.
- До момента зачатия врачи собирают семейный анамнез, опрашивая родителей об их родословной.
- Большое внимание специалисты уделяют факторам риска: возрасту матери, перенесенным инфекционным заболеваниям, хроническим болезням, вредным привычкам.
- Генетический анализ родителей – полноценное исследование, с помощью которого составляется их кариотип и обнаруживаются участки ДНК с дефектными генами.
Диагностика в период внутриутробного развития дает более точные результаты, поскольку обследуют организм плода. Пренатальная диагностика — важный этап в процессе выявления хромосомных нарушений.
- Ультразвуковое исследование плода и допплерография маточно-плацентарного кровотока – неинвазивные методы, полностью безопасные и рекомендованные всем беременным. Признаки синдрома Эдвардса: отставание плода в размерах и массе, большое количество околоплодных вод, видимые аномалии развития черепа и костей, агенезия пупочной артерии, малая величина плаценты, многоводие, брадикардия, отсутствие носовых костей, 2 артерии в пуповине, кисты сосудистых сплетений. Диагностика с помощью ультразвукового исследования является достоверной на 100%.
- Стандартный пренатальный скрининг включает анализ крови на сывороточные маркеры. Полученные результаты соотносят с возрастом беременной женщины и сроком гестации. При отклонении показателей от нормы ставят высокий риск синдрома Эдвардса. В таких случаях показано искусственное прерывание беременности по медицинским показаниям.
- Амниоцентез – клеточный анализ околоплодных вод. Инвазивная методика, осуществляемая путем забора амниотической жидкости шприцем. Ее клетки содержат образцы ДНК плода, которые проверяют на наличие генетических заболеваний.
- Кордоцентез — исследование пупочной крови плода, позволяющее определить генетические аномалии с высокой точностью.
- Биопсия хориона представляет собой пункцию матки через переднюю брюшную стенку и забор ткани для анализа – стандартного генетического исследования.
Беременным, попадающим в группу высокого риска, предлагается проведение инвазивной дородовой диагностики с последующим кариотипированием плода. Инвазивные методы считаются самыми точными и надежными, но требующими оперативного вмешательства и проникновения в оболочку плода. Диагноз подтверждается при помощи определения кариотипа малыша путем КФ-ПЦР.
Диагностика синдрома Эдвардса после рождения самая легкая, быстрая и точная. После выявления некоторых врожденных дефектов проводят генетический анализ для подтверждения диагноза. Основной задачей при рождении ребенка с этой патологией является обнаружение аномалий в развитии внутренних органов, которые обычно приводят к смерти в первые месяцы жизни. Именно на их поиск направлено большинство диагностических процедур непосредственно после рождения. Наиболее важными диагностическими исследованиями, которые должны быть выполнены ребенку с синдромом Эдвардса в первые часы жизни, являются эхокардиография, УЗИ органов брюшной полости и УЗИ почек.
Видео: синдром Эдвардса на УЗИ у плода, расширение воротниковой зоны
Лечение
Синдром Эдвардса, как и все хромосомные аномалии, неизлечим. Патология в большинстве случаев несовместима с жизнью. Больным детям показана симптоматическая терапия, направленная на поддержание физиологических функций организма, продление жизни и улучшение ее качества.
Коррекция патологических процессов, опасных для жизни, заключается в назначении противовоспалительного и антибактериального лечения при наличии пневмонии, восстановлении проходимости пищеварительного тракта при атрезии кишечника, кормлении через зонд, использовании пеногасителей при метеоризме, слабительных средств при запоре. Для поддержания жизнеспособности всего организма проводится коррекция работы дыхательной и пищеварительной систем, а также нормализация сердечной деятельности. Больным необходимо создать стерильные условия, чтобы избежать развития различных инфекционных заболеваний. Дети с данным синдромом требуют тщательного ухода и регулярного прохождения медицинского обследования.
Пупочные и паховые грыжи, пороки сердца и прочие аномалии лечат хирургическим путем, если состояние больного ребенка остается удовлетворительным. Оперативное вмешательство несет неоправданный риск. Хирурги, исправив внешние дефекты, могут потерять пациента из-за сбоя в работе сердечно-сосудистой системы и развития прочих серьезных осложнений.
Все дети с синдромом Эдвардса без исключения имеют отклонения в психофизическом развитии. Им требуется специальная программа воспитания, включающая комплекс обучающих методик. Она не восстановит нарушенные функции, но поможет привить некоторые элементарные бытовые навыки.
Прогноз и профилактика
Прогноз при синдроме Эдвардса крайне неблагоприятный. Больные дети погибают на первом году жизни. Только единицы доживают до юных и зрелых лет. При этом они имеют значительные умственные отклонения и нуждаются в постоянном уходе. Выжившие дети страдают от умственной отсталости и различных заболеваний, связанных с аномалиями строения. Только легкая форма мозаичного синдрома Эдвардса поддается коррекции. Больные дети начинают общаться с ограниченным кругом людей и приобретают элементарные навыки самообслуживания. Если обеспечить необходимый уход, ребенок может научиться самостоятельно поднимать голову и есть.
Чтобы предотвратить рождение больного ребенка, необходимо своевременно выявлять хромосомные патологии у плода. Для этого всем беременным женщинам не следует пренебрегать антенатальным скринингом.
В настоящее время в нашей стране работают специальные центры, в которых ухаживают за детьми с врожденными заболеваниями и развивают их интеллект. Если больной ребенок живет в таком центре больше года под наблюдением врачей, он начинает улыбаться, реагировать на движение, самостоятельно поддерживать положение тела и питаться.
Синдром Эдвардса – патология, которую фактически невозможно ни прогнозировать, ни лечить, ни корректировать. Данный диагноз является показанием к аборту.
Видео: о детях с синдромом Эдвардса
что это. Симптомы, причины, диагностика и лечение синдрома Эдвардса
Синдром Эдвардса представляет собой хромосомную патологию, которая характеризуется наличием дополнительной хромосомы (трисомия) в 18 паре.
Это тяжелая врожденная патология, вторая по частоте после синдрома Дауна, характеризующаяся тяжелыми аномалиями в развитии внутренних органов с неблагоприятным прогнозом. Согласно статистическим данным, частота болезни во всем мире находится в пределах 0,015—0,02%. На три-четыре больных девочки приходится один мальчик.
Причины синдрома Эдвардса
Синдром Эдвардса — это генетическое заболевание. Суть патологии заключается в наличии дополнительной третьей хромосомы в 18 паре.
Хромосомы — это очень компактно упакованная молекула ДНК, в которой закодированы все свойства человеческого организма. Каждая нормальная клетка содержит 46 хромосом. 44 из них являются соматическими, то есть, несут информацию о развитии и функционировании всех органов и систем, еще 2 — половые, определяющие принадлежность к тому или иному полу.
Так как синдром Эдвардса — это хромосомное заболевание, то в его основании лежит не мутация определенного гена, а дефект всей хромосомы, то есть, целой молекулы ДНК, а именно — дополнительная хромосома.
Точные причины синдрома Эдвардса не установлены. Обычно появление лишней хромосомы возможно до оплодотворения, что связано с аномалиями в генетическом материале сперматозоида или яйцеклетки. Провоцирует такое нарушение возраст родителей старше 40 лет, наличие подобных хромосомных нарушений в семье одного из родителей ребенка, а также злоупотребление алкоголем и курение одного из будущих родителей, прием лекарственных средств, ионизирующее излучение.
Вероятность развития хромосомных патологий напрямую зависит от возраста родителей, что доказано в ходе крупных научных исследований. Особенно важное значение имеет возраст матери — после 40 лет возможность зачать больного ребенка увеличивается примерно в 7 раз в сравнении с общей популяцией. Возраст отца также важен, но здесь зависимость выражена не так сильно.
Вредные вещества — этиловый спирт, сигаретный дым, наркотические соединения, при регулярном воздействии нарушают процессы созревания половых клеток и способствуют неправильному распределению хромосом. Как следствие рождается ребенок с тяжелой патологией.
Ионизирующие лучи, независимо от типа излучения, способны вызывать мутации в половых клетках. Особенно опасно влияние больших доз радиации в период полового созревания, когда в половые органы интенсивно растут и развиваются.
Узнайте экспертное мнение
Оставьте свой e-mail и мы расскажем, как правильно обследоваться и приступить к лечению
Признаки и симптомы синдрома Эдвардса
Симптомы синдрома Эдвардса во многом зависят от формы заболевания у конкретного больного.
На сегодняшний день генетики различают три типа болезни:
- полная трисомия;
- частичная трисомия;
- мозаичный синдром Эдвардса.
Полная трисомия — наиболее тяжелая форма, при которой симптомы синдрома Эдвардса выражены сильнее всего. В этом случае дополнительная хромосома содержится во всех клетках организма больного. Этой формой страдает до 90% больных.
Частичная трисомия диагностируется только у 3 больных из 100. В этой ситуации клетки содержат лишь фрагмент восемнадцатой хромосомы. Заболевания имеет место, если причиной патологии стали нарушения в процессе размножения половых клеток и, как следствие, неправильное распределение ДНК. Прогноз при частичной трисомии несколько лучше, чем при полной.
На мозаичную форму приходится до 7% случаев. В этом случае болезнь развивается после зачатия, при условии, что на плодное яйцо воздействовали неблагоприятные факторы. Именно в этом случае причины синдрома Эдвардса — химические вещества и физические факторы, поступающие извне. Как известно, уже в первые минуты после оплодотворения в яйцеклетке начинаются интенсивные процессы деления. Если в них произойдет сбой, то при очередном делении клетка получит дополнительную хромосому. В дальнейшем, она станет источником таких же дефектных клеточных элементов. Максимальное количество аномальных клеток при мозаичной форме синдрома Эдвардса — 50%.
Дети в случае наличия такого изменения количества хромосом рождаются со сниженной массой тела и наличием множественных пороков развития, которые заключаются в изменении формы черепа, уменьшении диаметра рта, аномалии твердого неба, наличии узких глазных щелей, деформации ушных раковин, дефекты конечностей, половых органов, специфические дерматоглифические проявления. Также часто развиваются пороки сердца, которые заключаются в нарушении заращения перегородок и формирования клапанов между его полостями. Благодаря яркой клинической картине диагностика заболевания не представляет серьезных трудностей.

Долихоцефалия — типичный признак заболевания. Череп больных детей имеет узкую и удлиненную форму, что подтверждают специальные измерения (соотношение ширины черепа к его длине). В норме показатель должен быть не менее 75%. Иногда эта форма встречается и у здоровых малышей, но если изменение заметно невооруженным глазом, без измерений, то вероятность того, что ребенку будет поставлен диагноз «синдром Эдвардса», очень велика.
Еще один внешний признак патологии — микроцефалия, при котором голова очень мала по отношению к туловищу. Особенно сильно недоразвита мозговая часть черепа.
У 95% больных детей наблюдается аномалия ушной раковины. Хрящ, который ее формирует, плоский. Выпуклости, характерные для здоровых людей, отсутствуют или выражены очень слабо, а сами раковины расположены низко. Нередко отсутствуют мочки уха или козелок. Слуховой ход сужен или атрезирован. Полное отсутствие слухового хода наблюдается у каждого 4 больного.
Еще одна характерная черта патологии — волчья пасть. Это врожденная аномалия развития твердого неба, которая появляется из-за несрастания небных отростков верхней челюсти.
В зависимости от степени выраженности различают 4 варианта порока:
- несрастание только мягкого неба;
- несрастание мягкого и части твердого неба;
- полное несрастание твердого неба;
- полное несрастание мягкого, твердого неба и губы.
Деформация стопы в виде стопы-качалки обусловлена неправильным расположением костей. При этом пятка сильно выступает назад, свод стопы отсутствует. Вогнутая линия, которая в норме соединяет пятку и большой палец, отсутствует, нередко стопа выпуклая, из-за чего похожа на ножки кресла-качалки, что и обусловило название деформации.
При наличии стопы-качалки часто наблюдается непропорциональное строение пальцев ног. Большой палец у больных детей короткий — короче, чем второй палец стопы. Важно понимать, что у взрослых этот признак не имеет диагностической ценности, так как может появиться из-за других заболеваний, неправильной обуви, суставной аномалии. Вместе с непропорциональным строением нередко имеет место сращение пальцев. В легких случаях это лишь кожная складка, но нередко обнаруживают и костные дефекты.
Кисти малыша находятся в специфическом положении — они сжаты в кулак, но мизинец и указательный палец при этом располагаются поверх среднего и безымянного. Это так называемое флексорное положение кисти, обусловленное повышенным тонусом мышц-сгибателей.
Недоразвитая нижняя челюсть встречается у 70% детей, которым поставлен диагноз «синдром Эдвардса». Подбородок сильно втянут, что заметно невооруженным глазом. Ребенок не может держать рот закрытым, что ведет к вытеканию слюны. У выживших детей формируется неправильный прикус.
Из аномалий половых органов чаще всего имеют место недоразвитие полового члена и увеличение клитора. Также возможно неправильное расположение уретры. У мальчиков яички могут оставаться в брюшной полости или паховом канале. В большинстве случаев эти отклонения сочетаются с пороками внутренних половых органов и мочевого пузыря.
Дерматоглифические симптомы — характерные отклонения в расположении узоров и складок на ладонях. В большинстве случаев дерматоглифика используется как предварительная диагностика синдрома Эдвардса, особенно частичной и мозаичной форм. При полной трисомии клиническая картина выражена настолько ярко, что потребность в детальном изучении узора ладоней отпадает за ненадобностью.
Важно понимать, что сами по себе вышеперечисленные признаки — далеко не диагноз. Единичные отклонения могут быть вариантом нормы у абсолютно здоровых людей, но наличие нескольких наиболее часто встречающихся отклонений является поводом для более детального обследования.
Диагностика синдрома Эдвардса
Диагностика синдрома Эдвардса заключается в генетическом исследовании, которое может проводиться во время беременности. При этом выполняется цитологическое исследование околоплодных вод с определением количества хромосом клеток эпидермиса плода. Так как подобные манипуляции провоцируют определенные осложнения, синдром Эдвардса с их помощью диагностируют только пациентам из группы риска.
Лечение синдрома Эдвардса
На данный момент лечение синдрома Эдвардса невозможно, так как хромосомная аномалия затрагивает все клетки плода или родившегося ребенка. Поэтому важно диагностировать синдром Эдвардса на ранних сроках течения беременности.
В случае подтверждения диагноза возможно проведение прерывания беременности по медицинским показаниям. Для родившегося ребенка с таким хромосомным нарушением прогноз неблагоприятный. Большинство детей не доживают до годовалого возраста. Выжившие дети с синдромом Эдвардса значительно отстают в развитии (особенно в умственном) и требуют постоянного медицинского наблюдения. В таком случае на первый план выходит тщательный уход и симптоматическое лечение синдрома Эдвардса.
Профилактика синдрома Эдвардса
Профилактика синдрома Эдвардса сводится к тщательному планированию предстоящей беременности. Всем парам из группы риска настоятельно рекомендуют проконсультироваться у генетика. Врач детально изучит семейный анамнез пары, выявит факторы риска, проведет генетический анализ пары.
При сборе семейного анамнеза доктор расспросит о наследственных заболеваниях в семье. При наличии таковых, вероятность рождения ребенка с аномалией возрастает до 1%. Очень желательно, чтобы родители собрали максимум информации о своих предках.
Затем генетик выявит факторы риска и даст рекомендации по их устранению, если это возможно.
Генетический анализ необходим для обнаружения дефектных генов в молекуле ДНК. Чем больше мутаций будет выявлено, тем выше вероятность рождения больного ребенка.
Всем беременным из группы риска показана пренатальная диагностика, благодаря которой можно вовремя диагностировать болезнь и принять решение по поводу дальнейшего вынашивания ребенка. В принципе, это также профилактика синдрома Эдвардса.
Из-за множественных пороков развития примерно половина детей умирает в первые три месяца жизни. Только 10 детей из 100 доживут до года. Более старшие дети нуждаются в тщательном уходе, постоянном лечении и профилактике осложнений синдрома Эдвардса. При мозаичной или частичной форме прогноз более благоприятный. Длительность жизни детей больше, но они также нуждаются в уходе. При полноценных реабилитационных мероприятиях малыш может научиться сидеть и питаться, иногда хорошо выражена реакция на окружающих. Из-за серьезной умственной отсталости больные не способны устроиться даже на простейшую работу.
Синдром Эдвардса можно предотвратить, если вы беременеете с помощью ЭКО. В клинике «АльтраВита» практикуется диагностическая процедура под названием ПГД (преимплантационная генетическая диагностика). Она дает возможность выявить хромосомную аномалию еще до помещения эмбриона в полость матки, то есть, до наступления беременности. Для переноса будет выбран тот эмбрион, который гарантированно не имеет никаких генных или хромосомных мутаций. Позвоните в клинику «АльтраВита», чтобы узнать о возможности проведения ПГД.
Синдром Эдвардса - Симптомы и лечение. Журнал Медикал
Синдром Эдвардса является одной из форм редкого генетического заболевания, когда часть 18-хромосомы человека дублируется. Большинство детей с данной патологией умирают еще на стадии эмбрионального развития, это происходит в 60 % случаев. Распространенность синдрома Эдвардса в среднем составляет 1:3000—1:8000 случаев. Наследование синдрома не прослеживается, а случайность данной мутации составляет всего 1%.
Синдром Эдвардса был назван в честь доктора Джона Эдварда, который в 1960 году описал первые случаи и зафиксировал закономерность развития симптомов. Синдром Эдвардса затрагивает больше женский пол, чем мужской - около 80 процентов пострадавших составляют женщины. Женщины старше тридцати лет имеют больший риск рождения ребенка с синдромом, хотя то же может происходить и с женщинами моложе тридцати, но значительно реже. Около 12% детей с синдромом доживают до возраста, когда можно оценить возможности умственного развития; младенцы, которые выживают, имеют серьезные дефекты и обычно живут не долго. Синдром Эдвардса связан с широким спектром нарушений, которые состоят более чем из ста тридцати дискретных дефектов, связанных с мозгом, сердцем, черепно-лицевой структурой, почками и желудком.
Причины развития синдрома Эдвардса.
Каждая клетка в организме человека содержит в себе двадцать три пары хромосом, которые он наследует от родителей. У людей в каждой половой клетке содержится одинаковое число хромосомных наборов. У женщин это яйцеклетки (их называют XX), а у мужчин (их называют XY) это сперматозоиды. Во время деления оплодотворённой яйцеклетки происходит мутация под воздействием каких-либо факторов и возникает дополнительная хромосома в восемнадцатой паре, которая и несет ответственность за возникновение синдрома Эдвардса. Дети с синдромом наследуют неправильное число хромосом, вместо двух копий у них образуется три копии хромосом. Данный вид мутации называют «трисомия», так же к названию прилагается номер пары, в которой произошла мутация, в случае синдрома Эдвардса это восемнадцатая пара.
Выше описанный вариант является «полной трисомией», т. е. ребенок унаследовал полную дополнительную копию лишней хромосомы, таких детей с синдромом Эдвардса по статистике 95%. «Полная трисомия» имеет практически все признаки заболевания и протекает очень тяжело.
Существует еще два варианта мутаций. Два процента детей с синдромом Эдвардса имеют транслокацию в 18 паре, когда присутствует только часть лишней хромосомы. Три процента детей синдромом имеют «мозаичную трисомию», данный вид мутации характеризуется наличием лишней хромосомы не во всех клетках организма.
Симптомы синдрома Эдвардса.
Большинство детей, рожденных с синдромом Эдвардса, имеют дефицит массы тела и выраженную задержку в развитии. Их голова необычно мала, а затылок имеет выраженный размер. Их уши низко посажены, верхняя и/или нижняя челюсть имеет дефект развития, называемый микрогнатией. Это состояние, когда искажается форма лица и формируется неправильный прикус.

Внешний вид больного с Синдромом Эдвардса
У детей часто формируется «волчья пасть» и «заячья губа», состояния, когда возникает расщелина в верхнем небе и губе соответственно.
Сформировавшаяся "заячья губа"
Ручки детей часто сжаты в кулачки и все пальцы имеют характерную неровную позицию.
Флексорное положение кисти
Дети страдают косолапостью, а пальцы на нижних конечностях могут иметь перепонки или полностью сливаться между собой.
Очень часто у больных с синдромом возникают проблемы с сердцем, легкими и диафрагмой из-за патологии сосудистого звена и развития врожденных пороков. Сердечные пороки могут иметь различный характер и различное количество: открытый артериальный проток, дефект межжелудочковой перегородки, открытое овальное отверстие.
Мутации могут вызывать развитие паховых и пупочных грыж, аномалии развития мочеполовой системы и диспластический синдром. Процентное соотношение развития того или иного порока:
Поражение системы и порок (признак) - Частота %
Мозговой череп и лицо - 100%
микрогения - 96,8%
низкое расположение или/и деформация ушных раковин - 95,6%
долихоцефалия - 89,8%
высокое небо - 78%
расщепленное небо - 15%
микростомия - 71%
Опорно-двигательный аппарат - 98,1%
флексорное положение кистей - 91,4%
стопа-качалка - 76,2%
кожная синдактилия стоп - 49,5%
косолапость - 34,9%
Центральная нервная система - 20,4%
Сердечно-сосудистая система - 90,4%
Органы пищеварения - 54,9%
Мочеполовая система - 33,5%
У детей с синдромом Эдварда, как правило, возникают проблемы с кормлением. Проблемы возникают из-за нарушения врожденных рефлексов глотания и сосания. Слабое развитие сосательного рефлекса и несогласованное глотание приводит к удушью ребенка.
Ребенок может заболеть гастроэозофагиальной рефлюксной болезнью. Врожденные расщелины лицевого скелета также вызывают трудности в скармливании. Они приводят к необходимости кормить ребенка через зонд или гастростому. Специалист может показать родителям ребенка, как правильно держать голову и тело своего ребенка. В целях предотвращения рефлюкса голова ребенка должна быть приподнята примерно на тридцать градусов или более пока ребенок ест, и в течение часа или двух после того как он поел.
Диагностика синдрома Эдварда
Диагноз синдрома Эдвардса может быть поставлен путем определения при физическом осмотре всех вышеперечисленных внешних симптомов. Более точный диагноз можно поставить, проведя анализ «кариотипирования», который включает в себя взятие образцов крови ребенка для определения хромосомного состава.
В связи с большим количеством пороков развития и очень низким процентом выживаемости детей с синдромом Эдвардса, в настоящее время разработаны методы антенатальной диагностики. Одной из первых и доступных методик, которые осуществляют во всех женских консультациях, является УЗИ плода. На УЗИ в ранних сроках беременности можно заподозрить пороки развития головного мозга и конечностей, также наличие обильного количество околоплодной жидкости, что должно насторожить врача. При получении данных результатов необходимо отправить беременную на более детальное и прицельное наблюдение в стационар.
В стационаре должны выполнить все необходимые общеклинические анализы и провести более специфические тесты, такие как эхография, доплерометрия, исследование сывороточных маркеров крови: β-субъединицы хорионического гонадотропина (βХГ), α-фетопротеина (АФП), эстриола (ЕЗ), 17-окси прогестерона. Для оценки степени риска рождения детей с хромосомной патологией во всех наблюдениях используют специально разработанную компьютерную программу PRISCA, учитывающую возраст женщины, показатели сывороточных маркеров и срок беременности. Также необходимо выполнить трансабдоминальный амниоцентез с последующим кордоцентезом. В амниотических водах при наличии синдрома Эдвардса будет определяться: АФП, 17-ОП, ЕЗ, а в крови плода будет определяться кариотип трисомии 18 хромосомы.
Лечение синдрома Эдвардса
Научному медицинскому сообществу не известно лекарство от синдрома в настоящее время. Дети с синдромом Эдвардса обычно рождаются с физическими отклонениями, и врачи сталкиваются с проблемой в отношении выбора метода лечения. Хирургия может устранить некоторые пороки, связанные с синдромом, но инвазивность методики не оправдывает результата у детей, чей срок жизни колеблется от нескольких дней до месяцев.
Лечение на сегодняшний день сводится к паллиативной помощи (поддержанию морального духа столкнувшихся со смертельным заболеванием). От пяти до десяти процентов детей с синдромом Эдвардса доживают до первого года жизни.
Проблемы, связанные с нарушениями нервной системы и тонуса мышц влияют на развитие двигательных навыков ребенка, что может привести к сколиозу, атрофии мышц, косоглазию. Хирургическое лечение у больных детей может быть ограничено из-за пороков развития сердечно-сосудистой системы. У детей с синдромом часто возникают запоры, вызванные плохим тонусом брюшной стенки и атоничным кишечником. В результате этого дети грудного возраста испытывают дискомфорт, возникают трудности с кормлением. Специальные молочные смеси, слабительные средства и препараты из группы «пеногасителей» могут облегчить данные симптомы. Клизмы в данной ситуации противопоказаны, потому что они могут привести к электролитному дисбалансу у ребенка.
Дети с синдромом часто отстают в развитие, в связи с этим необходимо применение специально разработанных программ терапии по коррекции задержки физического развития.
Еще одним фактором неблагоприятного прогноза является повышенный риск развития опухоли «Вильмса» – это одна из разновидностей рака почки. Рекомендуется регулярное УЗИ обследование брюшной полости на наличие данной патологии.
Больные дети подвержены риску развития различных инфекций и патологических состояний, таких как: инфекции мочеполовой системы, средний отит, конъюнктивиты различной этиологии, синуситы, фронтиты, пневмонии, ночное апноэ, легочная гипертензия, врожденные пороки сердца и повышенное артериальное давление. Необходимо быть готовыми, чтобы эффективно провести лечение и вовремя заподозрить данные заболевания.
Родителям, у которых ребенок имеет данный синдром, необходимо постоянное наблюдение за состоянием здоровья, так как своевременно выявленное заболевание может продлить жизнь их детям.
Прогноз синдрома Эдварда
Прогноз в большей своей степени при данной патологии не благоприятный. Большинство детей, которые родились с синдромом Эдвардса, не доживают до первого года своей жизни. Средняя продолжительность жизни для половины детей, рожденных с этим синдромом, менее чем два месяца. От девяноста до девяноста пяти процентов из этих детей умирает, не дожив до своего первого дня рождения. От пяти до десяти процентов детей, которые выжили в первый год, испытывают серьезные отклонениями в развитии.
Дети, которые прожили первый год, требуют постоянного контроля и наблюдения, и испытывают серьезные трудности в развитии. Их словесные навыки общения ограничены, хотя они в состоянии реагировать на утешения своих родителей и имеют возможность научиться улыбаться, распознавать и взаимодействовать с воспитателями и другими людьми. Они могут приобрести навыки, необходимые для всех детей, такие как самостоятельное кормление и поднятие головки, навыки, характерные для здоровых детей их возраста.
Врач терапевт Жумагазиев Е.Н.
Синдром Эдвардса - причины, риски, признаки на УЗИ
Большинство женщин хорошо знают, что такое синдром Дауна. И во время беременности многие узнают, что очень редко, но встречается и другое хромосомное нарушение, называемое Синдром Эдвардса. И многие обеспокоены, как узнать насколько высок риск рождения ребенка с синдромом Эдвардса и как диагностируется такая патология при беременности?
Что такое Синдром Эдвардса?
Синдром Эдвардса — это генетическое заболевание, характеризующееся дублированием (трисомией) ХVIII хромосомы и проявляющееся целым рядом характерных пороков развития у плода во время беременности, часто приводящих к смерти ребенка или его инвалидизации. То есть у ребенка вместо 46 хромосом образуется 47, эта лишняя хромосома дает другое название болезни — трисомия 18. Синдром был назван в честь исследователя Джона Эдвардса, который впервые описал его в 1960 году.
Почему возникает синдром Эдвардса — причины патологии
Даже если родители здоровы, и в семейном анамнезе нет такой патологии, ребенок с 18 хромосомой может родиться у любой женщины.
Как известно, в каждой человеческой клетке находится 46 хромосом, а в женских и мужских половых клетках по 23 хромосомы, которые, соединяясь при оплодотворении яйцеклетки, также дают в сумме 46 хромосом. Если говорить о синдроме Эдвардса, причины его появления неизвестны.
В настоящее время известно только то, что в результате определенных генетических мутаций появляется лишняя 47–я хромосома (дополнительная хромосома в 18–й паре хромосом, которых таким образом становится не 2, а 3). 
В 95% всех случаев развития синдрома Эдвардса в клетках находится именно лишняя 18–я хромосома (трисомия), однако в 2% наблюдается только «удлинение» 18–й хромосомы (транслокация), когда общее число хромосом остается нормальным и равняется 46.
В 3% случаев синдрома Эдвардса имеет место «мозаичная трисомия», когда дополнительная 47–я хромосома обнаруживается в организме не во всех клетках, а лишь определенной его части. Клинически все 3 варианта синдрома Эдвардса протекают практически одинаково, однако первый вариант может отличаться более тяжелым течением заболевания.
Как часто встречается эта патология?
Дети с синдромом Эдвардса погибают на этапе внутриутробного развития приблизительно в 60% случаев. Несмотря на это, среди генетических заболеваний данный синдром у выживших младенцев является достаточно распространенным, по частоте встречаемости уступая только синдрому Дауна. Распространенность синдрома Эдвардса составляет 1 случай на 3 — 8 тысяч детей.
Считается, что у девочек данное заболевание встречается в 3 раза чаще, а риск синдрома Эдвардса значительно возрастает, если беременной женщине 30 и более лет.
Смертность при синдроме Эдвардса на первом году жизни составляет около 90%, причем средняя продолжительность жизни при тяжелом течении заболевания у мальчиков — 2–3 месяца, а девочек — 10 месяцев, а до взрослого состояния доживают лишь единицы. Чаще всего дети с синдромом Эдвардса умирают от удушья, пневмонии, сердечно–сосудистой недостаточности или кишечной непроходимости — осложнений, вызванных врожденными пороками развития.
Как проявляется синдром у ребенка?
Признаки синдрома Эдвардса можно разделить на несколько больших групп:
К первой группе относятся симптомы, характерные для внешнего вида ребенка:
- При рождении низкая масса тела (около 2 100 – 2200 граммов)
- Непропорционально маленькая голова
- Дефекты развития верхней или нижней челюстей (микрогнатия)
- Искажение формы лица и формирование неправильного прикуса
- «Волчья пасть» (расщелина твердого неба) или «заячья губа» (расщелина верхней губы)
- Пальцы кисти сжаты, в кулаке располагаются неровно
- Низкая посадка ушей
- Перепончатость или полное слияние пальцев нижних конечностей
- Врожденная косолапость
- «Стопа–качалка»
- Относительно малых размеров ротовая щель (микростомия)
Ко второй группе можно отнести признаки нарушения работы внутренних органов, моторики и нервно–психического развития:
- Наличие врожденных пороков сердца, например, открытого овального отверстия, дефекта межжелудочковой перегородки, открытого артериального протока и т.п.
- Развитие паховых или пупочных грыж.
- Органы пищеварения: гастроэозофагиальная рефлюксная болезнь, нарушение глотательного и сосательного рефлекса, атрезия пищевода или заднего прохода, меккелев дивертикул, нарушение расположения кишечника.
- Центральная нервная система: задержка нервно–психического развития, умственная отсталость, недоразвитость мозжечка, мозолистого тела, сглаживание или атрофия мозговых извилин.
- Мочеполовая система: крипторхизм, гипоспадия у мальчиков, гипертрофия клитора, недоразвитость яичников у девочек, независимо от пола — подковообразная или сегментированная почка, удвоение мочеточников.
- Косоглазие, сколиоз, атрофия мышц.
Как узнать патологию во время беременности — диагностика
Синдром Патау, Эдвардса и другие трисомии лучше всего выявить до рождения малыша. Как правило, пренатальная диагностика данного синдрома проводится в 2 этапа:
- На сроке в 11–13 недель (скрининг, в основе которого — проведение различных биохимических анализов у женщины).
- Определение кариотипа плода у беременных их группы риска.
В 11–13 недель в крови женщины определяется уровень некоторых белков крови: β–ХГЧ (β–субъединица хорионического гормона человека) и плазменного протеина А ассоциированного с беременностью. Затем с учетом этих данных, возраста беременной женщины рассчитывается риск рождения ребенка с синдромом Эдвардса, и формируется группа риска беременных.
Далее в группе риска на более позднем сроке берется материал у плода для постановки точного диагноза: в 8–12 недель это биопсия ворсин хориона, в 14–18 — амниоцентез (изучение околоплодных вод), спустя 20 недель — кордоцентез (внутриутробное взятие крови из пуповины плода с УЗИ–контролем). После этого в полученном материале определяют наличие или отсутствие дополнительной 18–й хромосомы с помощью КФ–ПЦР (количественной флуоресцентной полимеразной цепной реакции).
Если беременная не прошла генетическое скрининг–обследование, то на более поздних сроках предварительная диагностика синдрома Эдвардса осуществляется с помощью УЗИ. Прочие косвенные признаки, на основании которых можно заподозрить синдром Эдвардса на более поздних сроках:
- Наличие аномалий развития костей и мягких тканей головы («волчья пасть», микроцефалия, низкая посадка ушных раковин, «заячья губа» и т.п.).
- Обнаружение пороков со стороны сердечно–сосудистой , мочеполовой системы, а также опорно–двигательного аппарата.
Диагностические признаки синдрома у ребенка
После рождения ребенка опорными диагностическими признаками наличия синдрома Эдвардса являются следующие:
- Микроцефалия, малый вес при рождении
- Наличие «заячьей губы» или «волчьей пасти»
Признаки характерной дерматографической картины:
- неразвитая на пальцах дистальная сгибательная складка
- наличие в 1/3 случаев поперечной ладонной борозды
- дуги на подушечках пальцев рук
- изменение кожного рисунка ладони: дистальное расположение осевого трирадиуса и увеличение гребневого счета.
Далее диагноз также подтверждается с помощью определения кариотипа ребенка методом КФ–ПЦР.
Синдром Эдвардса на УЗИ — кисты сосудистых сплетений
На ранних сроках синдром Эдвардса на УЗИ заподозрить крайне трудно, однако в 12 недель беременности уже выявляют характерные для него симптомы косвенного характера:
- Признаки задержки развития плода
- Брадикардия (снижение у плода частоты сердечных сокращений)
- Омфалоцеле (наличие грыжи брюшной полости)
- Отсутствие визуализации косточек носа
- В пуповине одна, а не 2 артерии
Также на УЗИ могут быть обнаружены кисты сосудистых сплетений, представляющих собой полости с содержащейся в них жидкостью. Сами по себе они не несут угрозу для здоровья и исчезают к 26–недельному сроку беременности. Однако такие кисты очень часто сопровождают различные генетические заболевания, например, синдром Эдвардса (в данном случае кисты обнаруживаются у 1/3 детей, страдающих этой патологией), поэтому при обнаружении таких кист врач направит беременную женщину на обследование в генетическую консультацию.
Лечение
Так как дети с синдромом Эдвардса редко доживают до года, то сначала лечение направлено на коррекцию тех пороков развития, которые опасны для жизни:
- восстановление прохода пищи при атрезии кишечника или анального отверстия
- кормление через зонд в случае отсутствия глотательного и сосательного рефлексов
- антибактериальная и противовоспалительная терапия при воспалении легких
При относительно благоприятном течении заболевания происходит коррекция некоторых аномалий и пороков развития: хирургическое лечение «волчьей пасти», пороков сердца, паховой или пупочной грыжи, а также симптоматическое медикаментозное лечение (назначение слабительных при запорах, «пеногасителей» при метеоризме и т.п.). 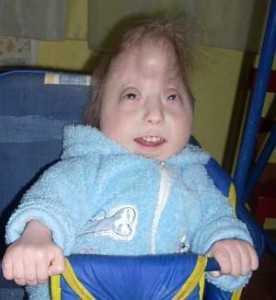
Дети с синдромом Эдвардса склонны к таким заболеваниям, как:
- средний отит
- рак почки (опухоль Вильмса)
- пневмония
- конъюнктивит
- легочная гипертензия
- апноэ
- повышенное артериальное давление
- фронтиты, синуситы
- инфекции мочеполовой системы
Поэтому лечение больных с синдромом Эдвардса включает своевременную диагностику и терапию данных заболеваний.
Прогноз для ребенка
В большинстве случаев прогноз неблагоприятный. Единицы детей с синдромом Эдвардса, которые доживают до взрослого возраста, имеют серьезные умственные отклонения и постоянно требуют постороннего ухода и контроля. Однако при соответствующих занятиях они способны реагировать на утешение, улыбаться, самостоятельно принимать пищу, а также взаимодействовать с воспитателями, приобретая различные умения и навыки.
Синдром Эдвардса: фото, причины, диагностика, лечение
Сегодня мы поговорим о достаточно редком детском заболевании, сопровождающемся большим числом аномалий и нарушений в развитии. Речь пойдет о синдроме Эдвардса. Разберем его причины, формы, частоту проявления, методы диагностики и прочие важные вопросы.
Что это?
Синдром Эдвардса - болезнь, обусловленная хромосомными аномалиями, вызывающая целый список нарушений и отклонений в развитии ребенка. Ее причина - трисомия 18-й хромосомы, т. е. присутствие ее лишней копии. Этот факт и ведет к появлению осложнений генетической природы.
Риск того, что родится ребенок с синдромом Эдвардса, равен 1 случаю из 7000. К сожалению, большинство младенцев с данным отклонением умирают в первые недели жизни. Только порядка 10 % живут один год. Болезнь ведет за собой глубокую умственную отсталость, врожденные поражения внутренних и внешних органов. Самые частые из них - порок мозга, сердца, почек, маленькая голова и челюсть, расщелина губы или неба, косолапость.

Впервые сформированы и описаны симптомы заболевания были в 1960 году Д. Эдвардсом. Доктор сумел установить зависимость между проявлением нескольких признаков, обнаружил более 130 дефектов, которые сопутствуют заболеванию. Хоть симптомы синдрома Эдвардса проявляются весьма ярко, современные методы терапии против них, к сожалению, бессильны.
Причина заболевания
Если синдром Эдвардса (фото больных детей по этическим соображениям не будут размещены) был диагностирован во время беременности, то чаще всего последняя заканчивается выкидышем или мертворождением. Увы, проявление болезни у плода предотвратить сегодня нельзя.
Также в современности не выяснены четкие причины этого генетического заболевания, отчего и профилактические меры против развития его у будущих детей сформировать нельзя. Однако специалисты определили факторы риска:
- Неблагоприятные экологические условия.
- Воздействие радиации, токсических, химических веществ на родителей.
- Пристрастие к алкоголю и табаку.
- Наследственность.
- Прием определенных медикаментов.
- Инцест, кровное родство родителей.
- Возраст будущей матери. Если женщине старше 35 лет, то это считается причиной синдрома Эдвардса у ребенка, равно как и иных хромосомных заболеваний.
Формы синдрома
На тип такой аномалии в первую очередь влияет стадия развития зародыша, на которой синдром настигает эмбрион. Всего различаются три вида:
- Полный. Самый тяжелый тип, на него приходится 80 % случаев. Утроенная хромосома появляется в тот момент, когда плод был всего одной клеткой. Отсюда следует, что аномальный хромосомный набор будет передаваться при делении и всем остальным клеткам, наблюдаться в каждой из них.
- Мозаичный. Название дано из-за того, что здоровые и мутировавшие клетки смешаны, как мозаика. 10 % пораженных симптомом Эдвардса страдают именно этой формой. Признаки болезни здесь выявлены слабее, но все же мешают нормальному развитию ребенка. Избыточная хромосома появляется во время фазы, когда зародыш состоит из нескольких клеток, поэтому поражается только часть организма или отдельный орган.
- Возможная транслокация. Здесь наблюдается не только нерасхождение хромосом, но и переизбыток информации, порожденный транслокационной перестройкой. Проявляется как во время созревания гамет, так и во время развития зародыша. Отклонения здесь не ярко выражены.
Распространенность синдрома
Риск синдрома Эдвардса невозможно выразить в точных цифрах. Нижняя граница рождения ребенка с такой аномалией - 1:10000, верхняя - 1:3300. При этом встречается он в 10 раз реже, чем синдром Дауна. Средние показатели зачатия детей с болезнью Эдвардса выше - 1:3000.
Согласно исследованиям, риск родить малыша с таким синдромом повышается при возрасте родителей более 45 лет на 0,7 %. Но он и присутствует и у 20-, 25-, 30-летних родителей. Средний возраст отца ребенка с синдромом Эдвардса - 35 лет, матери - 32,5 лет.
Аномалия также связана с полом. Доказано, что у девочек она встречается в 3 раза чаще, чем у мальчиков.
Синдром и беременность
Проявляет свои признаки синдром Эдвардса еще на стадии беременности. Последняя протекает с рядом осложнений, характерна перенашиванием - малыши рождаются примерно на 42-й неделе.
На этапе беременности заболевание плода характеризует следующее:
- Недостаточная активность эмбриона.
- Брадикардия - сниженная частота сердечных сокращений.
- Многоводие.
- Несоответствие размера плаценты размеру плода - она имеет меньший размер.
- Развитие одной пупочной артерии вместо двух, что ведет к кислородной недостаточности, асфиксии.
- Грыжа брюшной полости.
- Сплетение сосудистых образований, видимое на УЗИ (обнаруживаются у 30 % детей, пораженных синдромом).
- Недостаточный вес плода.
- Гипотрофия - хроническое расстройство функций ЖКТ.
60 % детей погибают уже в материнской утробе.
Дородовая диагностика
Синдром Эдвардса на УЗИ возможно определить только по косвенным признакам. Самый точный метод диагностики синдрома у плода сегодня - перинатальный скрининг. На его основе при возникновении тревожных подозрений доктор уже направляет женщину на инвазивное тестирование.
Скрининг, выявляющий кариотип синдрома Эдвардса, разделяется на два этапа:
- Первый проводится на 11-13 неделе беременности. Исследуются биохимические показатели - проверяется кровь матери на уровень гормонов. Результаты на этом этапе не окончательны - они могут говорить только о наличии риска. Для расчетов специалисту нужен белок А, ХГЧ, белок, вырабатываемый оболочками эмбриона и плаценты.
- Второй этап уже направлен на точный результат. Для исследований берется образец пуповинной крови или околоплодной жидкости, который затем повергается генетическому анализу.

Инвазивное тестирование
Хромосомы синдрома Эдвардса вероятнее всего определить данным методом. Однако он обязательно предполагает оперативное вмешательство и проникновение в оболочки эмбриона. Отсюда риск прерывания беременности и развития осложнений, отчего тест назначается только в крайних случаях.
На сегодня известны три типа взятия образца:
- БВХ (биопсия ворсин хориона). Основное преимущество метода - образец берется, начиная с 8-й недели беременности, что позволяет на ранних сроках определить осложнения. Для исследований нужен образец хориона (один из слоев оболочки плаценты), структура которого схожа со структурой эмбриона. Данный материал позволяет диагностировать внутриутробные инфекции, генетические и хромосомные болезни.
- Амниоцентез. Анализ проводится, начиная с 14-й недели беременности. В этом случае зондом протыкаются амниотические оболочки эмбриона, инструмент собирает образец околоплодных вод, содержащих в себе клетки будущего ребенка. Риск развития осложнений от такого исследования гораздо выше, чем в предыдущем случае.
- Кордоцентез. Срок - не ранее 20-й недели. Здесь берется образец пуповинной крови эмбриона. Сложность в том, что при взятии материала у специалиста нет права на ошибку - он должен попасть иглой точно в сосуд пуповины. На практике это происходит так: через переднюю стенку брюшины женщины вводится пункционная игла, которая собирает порядка 5 мл крови. Процедура проходит под контролем УЗИ-устройств.
Все вышеописанные методы нельзя назвать безболезненными и безопасными. Поэтому их проводят только в случаях, когда риск генетического заболевания у плода выше риска развития осложнений от взятия материала для анализа.
Родителям надо помнить, что ошибка медика при процедуре может привести к проявлению серьезных заболеваний, врожденных пороков у будущего ребенка. Нельзя исключить и риск внезапного прерывания беременности от подобного вмешательства.
Неинвазивное тестирование
Диагностика синдрома Эдвардса у плода включает в себя и неинвазивные методы. То есть без проникновения в плодные оболочки. Притом в точности такие методы не уступают инвазивным.
Одним из высокоточных анализов данного типа можно назвать кариотипирование. Это взятие образца крови матери, содержащей в себе свободные ДНК эмбриона. Специалисты извлекают их из материала, копируют, после чего проводят необходимые исследования.
Послеродовое диагностирование
Специалист может определить детей с синдромом Эдвардса и по внешним признакам. Однако для постановке окончательного диагноза проводятся следующие процедуры:
- УЗИ - исследование патологий внутренних органов, обязательно сердца.
- Томография головного мозга.
- Консультация детского хирурга.
- Обследования у специалистов - эндокринолога, невролога, отоларинголога, офтальмолога, ранее работавших с детьми, страдающими от данного заболевания.
Отклонения при синдроме
Патологии, причина которых - трисомия по 18 хромосоме, довольно серьезны. Поэтому только 10 % детей с синдромом Эдвардса доживают до года. В основном же девочки живут не более 280 суток, мальчики - не более 60.
У детей наблюдаются следующие внешние отклонения:
- Вытянутый от макушки к подбородку череп.
- Микроцефалия (маленький размер головы и мозга).
- Гидроцефалия (скопление жидкости в черепной коробке).
- Узкий лоб при широком затылке.
- Аномально низкое расположение ушей. Может отсутствовать мочка или козелок.
- Укороченная верхняя губа, делающая рот треугольным.
- Высокое небо, часто со щелью.
- Деформированные челюстные кости - нижняя челюсть аномально маленькая, узкая и неразвитая.
- Укроченная шея.
- Аномально узкие и короткие глазные щели.
- Отсутствие части глазной оболочки, катаракта, колобома.
- Нарушение функций суставов.
- Недоразвитые, малоподвижные стопы.
- Из-за аномального строения пальцев могут формироваться ластообразные конечности.
- Порок сердца.
- Ненормально расширенная грудная клетка.
- Нарушенная работа эндокринной системы, в частности, надпочечников и щитовидной железы.
- Необычное расположение кишечника.
- Неправильная форма почек.
- Удвоение мочеточника.
- У мальчиков - крипторхизм, у девочек - гипертрофированный клитор.
Отклонения психического характера обычно следующие:
- Недостаточно развитый головной мозг.
- Осложненная степень олигофрении.
- Судорожный синдром.

Прогноз для больных синдромом Эдвардса
К сожалению, на сегодня прогнозы неутешительны - порядка 95 % детей с данной болезнью не доживают до 12 месяцев. При этом тяжесть ее формы не зависит от соотношения больных и здоровых клеток. У выживших детей отмечаются отклонения физической природы, тяжелая степень олигофрении. Жизнедеятельность такого ребенка нуждается во всестороннем контроле и поддержке.
Нередко дети с синдромом Эдвардса (фото не представлены в статье из этических соображений) начинают различать эмоции окружающих, улыбаться. Но их общение, умственное развитие при этом ограничено. Ребенок со временем может научиться сам поднимать голову, кушать.
Возможности терапии
Сегодня такое генетическое заболевание неизлечимо. Ребенку назначается только поддерживающая его состояние терапия. Жизнь пациента связана со многими аномалиями и осложнениями:
- Мышечная атрофия.
- Косоглазие.
- Сколиоз.
- Сбои в работе сердечно-сосудистой системы.
- Атония кишечника.
- Низкий тонус стенок брюшины.
- Отит.
- Пневмония.
- Конъюнктивит.
- Синусит.
- Заболевания мочеполовой системы.
- Большая вероятность развития рака почки.
Заключение
Подводя итог, хочется отметить, что синдром Эдвардса не передается по наследству. Больные в большинстве случаев не доживают до репродуктивного возраста. При этом они не способны к продолжению рода - для заболевания характерна недоразвитость половой системы. Что касается родителей ребенка с синдромом Эдвардса, то шанс постановки такого же диагноза при следующей беременности равен 0,01 %. Надо сказать, что и само заболевание проявляет себя весьма редко - диагностируется только у 1% новорожденных. Нет и особых причин для его возникновения - в большей части случаев родители совершенно здоровы.
Синдром Эдвардса: признаки, причины и диагностика
Внешние и внутренние проявления заболевания могут существенно различаться в зависимости от особенностей развития плода. Чаще всего хромосомные нарушения появляются на начальных этапах развития эмбриона, поэтому они сказываются на развитии всего организма. Есть несколько внешних признаков, по которым с большой вероятностью можно предположить наличие синдрома Эдвардса у новорожденных.
Одной из наиболее характерных черт этого заболевания является искаженная форма черепных костей: череп вытянут от макушки к подбородку, вместе с тем часто ставится диагноз «микроцефалия» (уменьшение размеров черепа и мозга) или «гидроцефалия» (скопление жидкости в головном мозге). Лоб узкий, а затылочная сторона более широкая и выступающая, при этом уши расположены ниже, чем при нормальном развитии. Деформируются челюстные кости – часто это приводит к значительному уменьшению нижней челюсти, она становится узкой и недоразвитой. Вследствие этого рот также небольшой и часто треугольный из-за укорочения верхней губы. Небо высокое, иногда присутствует щель. Шея может быть укороченной, с характерной складкой.
Глазные щели уже и короче, чем нужно, переносица расширена и вдавлена – это особенно заметно при том, что нос, как правило, заужен, косточки носа могут визуально отсутствовать. Глазное яблоко также подвержено изменениям и нарушениям, которые приводят к катаракте и колобоме, то есть отсутствию части глазной оболочки. Кроме того, могут быть и иные нарушения зрения.
Уши низко посажены и деформированы, часто в горизонтальной плоскости. На ухе часто отсутствует мочка, а иногда козелок. Наружный слуховой проход часто сужен, иногда может отсутствовать полностью.
Широкий спектр нарушений касается костного скелета. Прежде всего, суставы не могут функционировать нормально, поэтому стопы и кисти не могут сгибаться и разгибаться так, как нужно. К тому же стопы недоразвиты, из-за этого изменяется их форма, они становятся менее подвижными. Большой палец укорочен, а второй и третий срастаются, иногда настолько сильно, иногда формируются ластообразные конечности. В 80% случаев формируется стопа с провисающим сводом, выступающей пяткой и коротким большим пальцем.
Из-за излишней подвижности тазобедренного сустава нередко случаются вывихи.
На подушечках пальцев количество дуг может быть в 10 раз больше нормы, однако сгибательной складки пальцы не имеют. Почти у 30% больных на ладонях появляются поперечные борозды и множество гребешков.
Помимо всего прочего, при синдроме Эдвардса деформируется форма грудной клетки – она расширяется, а межреберные промежутки уменьшаются, таким образом она становится короче и шире.
Значительные изменения претерпевают и внутренние органы. Почти все пациенты имеют порок сердца. Как правило, он характеризуется недостаточной развитостью клапанов в артериях и аортах. При этом довольно часто появляется дефект межжелудочковой перегородки.
Имеют место очень серьезные нарушения в обменных процессах, таких как работа эндокринной системы. Вследствие хромосомных нарушений железы не могут функционировать нормально, поэтому рост существенно замедляется. Гормональные нарушения приводят к недоразвитости подкожной клетчатки. У каждого десятого отмечается нарушение работы надпочечников или щитовидной железы.
Пониженный мышечный тонус со временем обычно повышается, при этом улучшается кровообращение.
Примерно у половины больных наблюдается аномальное развитие кишечника. Чаще всего эта аномалия заключается в его необычном расположении, при этом появляется мешок, образованный из слоев стенки кишки, а пищевод сужается слишком резко. Почки часто бывают сегментированы или имеют неправильную форму дуги, также может быть удвоение мочеточников.
Изменения также затрагивают и половые органы. У мальчиков яичко может не опускаться в мошонку (крипторхизм) и меняется строение пениса. У девочек формируется гипертрофированный клитор, а яичники развиты недостаточно.
В целом картина внешних и внутренних отклонений при синдроме Эдвардса выглядит следующим образом. В 100% случаев наблюдаются аномалии строения черепа и изменение формы лица. Почти у 97% уменьшение челюсти (микрогения), чуть более чем в 95 % случаев нарушается строение и расположение ушных раковин. Удлинение черепа наблюдается почти у 90% пациентов, высокое небо – у 78%, а уменьшенный рот – в 71% случаев.
Что касается нарушений конечностей, то они есть у 98% больных. Чаще всего встречается изменение формы кистей (более 91%) и стоп (76%).
Развитие сердечно-сосудистой системы нарушено более чем у 90% пациентов. Около 1/3 пациентов имеют нарушения мочеполовой системы, а 55% –пищеварительной системы.
причины, симптомы, диагностика, лечение, профилактика
Трисомия 18 представляет собой наследственное заболевание, обусловленное множественными нарушениями в развитии органов и систем ребенка.
Причины
Причина развития патологии связана с наличием трех хромосом в 18 паре вместо двух. Возникновению патологии способствует проживание в районах с неблагоприятной экологической ситуацией, близкородственные браки, пожилой возраст беременной женщины, неблагоприятный семейный анамнез.
Симптомы
Нарушение может сопровождаться развитием различной симптоматики, свидетельствующей о поражении различных органов либо систем.
Со стороны опорно-двигательной системы у таких больных довольно часто определяются дефекты развития нижней челюсти, аномалии формирования черепной коробки, может наблюдаться удлинение черепа, деформация стоп, сужение глазной щели, слабое развитие скелетной мускулатуры и слабое развитие подкожно-жировой клетчатки.
При дефектах развития внутренних органов у больных могут выявляться различные пороки сердца, сосудов, дефекты пищевода, закрытие протоков желчевыводящих путей, нетипичное расположение внутренних органов, недоразвитие половых органов, неопущение у мальчиков яичек в мошонку, а у девочек – удвоение половых органов.
У таких больных может наблюдаться тяжелая задержка психоэмоционального либо физического развития, а также олигофрения.
Также у таких пациентов наблюдаться развитие симптомов поражения центральной нервной системы, таких как уменьшение объемов головного мозга, отсутствие либо недоразвитие определенных мозговых элементов.
Довольно часто дети с таким нарушением страдают уменьшением размера обоих глаз.
Диагностика
Наличие недуга можно предположит до родов. На него указывают определенные изменения в биохимических анализах крови женщины вынашивающей ребенка. Также косвенно указывать на наличие у плода патологии может тяжело протекающая беременность.
При наличии у пациентки других признаков генетической патологии плода, потребуется консультация специалиста-генетика.
При тяжелых нарушениях в развитии плода рассматривается вопрос об искусственном прерывании беременности.
Болезнь может быть диагностирована сразу же после рождения ребенка. Типичное сочетание некоторых характерных признаков позволяет заподозрить у ребенка наличие синдрома. Для постановки диагноза потребуется выполнение кариотипирования. Такое диагностирование может проводиться даже уже после смерти малыша, так как полученные во время такого исследования сведения позволят установить вероятность возникновения генетических недугов у других детей пары.
Дополнительно могут использоваться ультразвуковое изучение сердца и внутренних органов ребенка, компьютерная томография, рентгенография, а также другие инструментальные методы, выбранные по решению врача.
Лечение
Терапия недуга основана на организации качественного ухода за больным ребенком и выполнение всех рекомендаций врача. При необходимости таким детям может потребоваться проведение хирургического лечения или пластической коррекции дефектов.
Профилактика
На данный момент не разработано специальных методов лечения этого заболевания. При наличии неблагоприятного семейного анамнеза у одного из супругов, перед тем как будет принято решение о беременности, потребуется консультация генетика.
причины, характерные симптомы и эффективность лечения
Синдром Эдвардса – генетическое заболевание, возникающее на фоне трисомии 18 хромосомы. Клинически аномалия проявляется дефектами строения различных органов и систем: сердца, желудочно-кишечного тракта, скелета и суставов. Диагностика недуга предполагает проведение УЗИ во время беременности, а также использование генетического анализа ДНК родителей и плода. Специфического лечения патологии не разработано. Прогноз при выявлении заболевания неблагоприятный: большинство малышей погибают в первые месяцы жизни.
Причины появления синдрома Эдвардса
Патология имеет генетическую природу. Возникновение специфических симптомов связано с врожденными аномалиями строения организма ребенка. Дефекты, характерные для синдрома Эдвардса, возникают в результате дублирования 18 хромосомы как во время оплодотворения, так и после него. Мутации не свойственно наследование, поэтому семейный анамнез пациентов не осложнен выявлением подобной проблемы. Это связано с небольшой продолжительностью жизни детей, у которых возникает данная аномалия. В результате нарушения в процессе деления клеток образуется лишняя 47 хромосома, наличие которой и обеспечивает возникновение расстройства. При этом точные причины формирования синдрома Эдвардса на сегодняшний день неизвестны. Доказано, что риск развития генетической патологии повышается по мере увеличения возраста будущей матери. Период беременности у женщин старше 35 лет чаще сопряжен с возникновением различных осложнений.
В отдельных случаях врачи отмечают отсутствие трисомии 18 хромосомы. Регистрируется лишь удлинение данной структуры, то есть изменение ее строения, при этом суммарное количество соединений остается нормальным. Подобный вариант кариотипа диагностируется только у 2% пациентов с синдромом Эдвардса. В 3% случаев возникает мозаичная трисомия. Подтверждение наличия данной патологии наиболее проблематично. Это состояние сопряжено с присутствием дополнительной хромосомы в отдельных клетках организма. Признаки, характерные для синдрома Эдвардса, во всех случаях сходны. Однако возможна вариабельность по интенсивности проявления поражения.
Большинство малышей, страдающих от этой болезни, погибает еще до рождения. При этом даже с учетом данной статистики патология имеет широкое распространение. По частоте встречаемости она уступает только синдрому Дауна. Заболевание регистрируется у 1 новорожденного из 5–7 тысяч. Три четверти пациентов составляют девочки. Врачи связывают данную особенность с высоким процентом внутриутробной смертности у мальчиков.
Характерные симптомы
Первые признаки наличия заболевания выявляются еще до рождения ребенка. Они включают в себя неактивность плода, многоводие, а также уменьшение размеров плаценты. Зачастую у женщин происходит самопроизвольный аборт. Малыши с синдромом Эдвардса появляются на свет с недостаточной массой тела и другими симптомами недоношенности. Дети имеют неправильную форму черепа, зачастую удлиненную к затылку, страдают от гидроцефалии. Такие патологии, как «заячья губа» и «волчья пасть», сопровождающиеся формированием расщелин в области рта и неба, – распространенное проявление недуга. Принято различать несколько основных групп симптомов, регистрируемых при синдроме:
- Со стороны опорно-двигательного аппарата отмечается нарушение строения и функции суставов. У многих новорожденных конечности искривлены, неспособны к физиологическим движениям. Грудная клетка имеет неправильную форму, она расширена и укорочена. У некоторых детей отмечается сращение пальцев на ногах.
- Распространенные и кардиологические симптомы. Наиболее опасно формирование пороков сердца, таких как незаращение артериального протока, а также дефекты межжелудочковой перегородки.
- Эндокринная система претерпевает ряд патологических изменений. Вследствие этого дети имеют сниженную массу тела, значительно отстают по темпу роста от своих сверстников. Подкожная клетчатка сформирована плохо, поэтому зачастую новорожденные выглядят истощенными. Отмечается недоразвитие вилочковой железы и семенников, крипторхизм.
- Дефекты со стороны желудочно-кишечного тракта связаны с аномалиями формирования пищеварительной трубки. Регистрируется развитие атрезии различных участков ЖКТ. У малышей слабо выражен сосательный рефлекс, диагностируется гастроэзофагеальный рефлюкс.
- Дети с синдромом Эдвардса зачастую страдают от нарушений зрения. Это связано как с патологией развития центров в головном мозге, так и с неправильным строением черепа, деформациями хрусталика и связок глаз.
- У всех малышей присутствуют признаки умственной отсталости. Интенсивность данного симптома варьируется и зависит от типа хромосомного дефекта, выявляемого при заболевании.
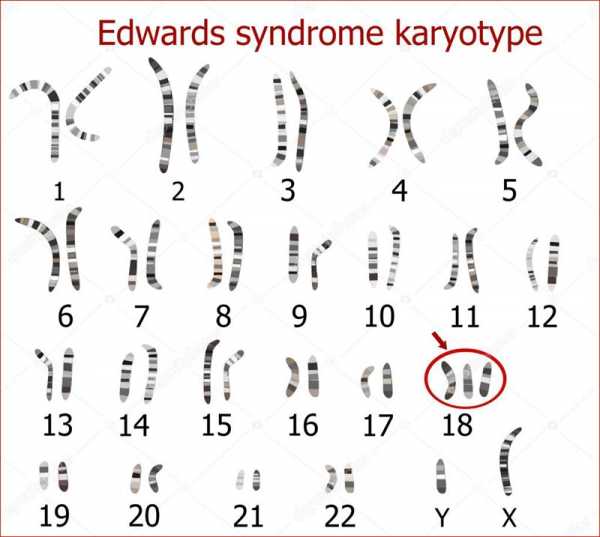
Необходимая диагностика
Подтверждение наличия проблемы возможно еще до рождения ребенка. Для этого используется пренатальный скрининг, основным методом которого является УЗИ. Фото, получаемые в результате проведения исследования, позволяют обнаружить косвенные признаки, указывающие на данный диагноз. По мере роста и развития плода симптомов становится все больше. Если во время беременности у врача появляется подозрение на синдром Эдвардса, будущая мать должна принять решение: рожать или нет. Для диагностики патологии используются и более инвазивные методики, включающие в себя проведение биопсии. После появления малыша на свет подтверждение наличия заболевания основано на клинических признаках и проведении кариотипирования.
У беременных
УЗИ проводится уже со срока 12 недель. В ходе исследования обнаруживаются такие аномалии:
- Увеличение количества околоплодной жидкости.
- Урежение пульса малыша.
- Отсутствие у ребенка носовых костей черепа.
- Формирование кист в местах закладывания крупных сосудистых сплетений.
Более информативно проведение УЗИ на поздних сроках, то есть в последнем триместре. В ходе осуществления диагностики врачи регистрируют дефекты строения черепа, пороки сердца и аномалии развития мягких тканей различной локализации. В процессе диагностики применяется и биохимический анализ крови, который проводится с 11 по 13 неделю беременности. Оценивается уровень хорионического гормона и плазменного протеина А. При синдроме Эдвардса концентрация этих соединений находится за пределами физиологической нормы. Несмотря на то что подобные тесты не позволяют поставить точный диагноз, риск выявления патологии в дальнейшем значительно повышается. Инвазивные методики включают в себя биопсию хорионических ворсин, забор амниотической жидкости и пуповинной крови. Однако подобные техники не применяются широко в связи с частыми осложнениями в виде инфицирования плода и абортов. Наиболее точный тест – анализ ДНК малыша и матери.

У новорожденных
Появление на свет ребенка с признаками недоношенности, а также дефектами развития различных систем указывает на синдром Эдвардса. Для постановки точного диагноза проводится кариотипирование. Это исследование позволяет определить хромосомный набор новорожденного, выявить в нем патогномоничные изменения.
Основную роль в подтверждении наличия генетической проблемы на сегодняшний день играет пренатальная диагностика, которая основана на регулярном скрининге показателей здоровья будущей матери, а также на обнаружении сонографических отклонения в развитии плода при помощи ультразвука. Однако клинические признаки, оцениваемые после появления ребенка на свет, также играют важную роль. В ряде случаев при синдроме Эдвардса у детей отмечаются лишь незначительные аномалии, которые сопряжены с более благоприятным прогнозом. Такие дефекты включают в себя маленькие ногти, недоразвитые пальцы на руках и ногах, а также короткую грудную клетку. Однако распространенными при недуге все же остаются врожденные пороки развития сердца и почек.
Несмотря на изученность патологии и известную ожидаемую смертность только 5–10% пациентов с синдромом Эдвардса живут дольше года. Основные причины гибели детей с данным заболеванием – это апноэ, респираторная и сердечная недостаточность. Остановка дыхания возникает как вследствие врожденных кардиальных пороков, так и на фоне аспирации или обструкции из-за нарушения функции желудочно-кишечного тракта, что должно быть учтено при оказании помощи, особенно в домашних условиях.
Эффективность лечения и прогноз
Специфического метода борьбы с патологией на сегодняшний день нет. Подобная проблема связана с генетической основой заболевания. Поэтому прогноз при синдроме Эдвардса неутешительный. Больше половины детей погибают в первые три месяца после рождения. Лишь малой части, не более 10%, удается дожить до года. Даже если получается спасти пациента, растить его очень сложно. Это связано с высоким риском формирования осложнений, а также выраженной умственной отсталостью, которая с трудом поддается коррекции. Исключение составляют лишь дети с мозаичным кариотипом синдрома Эдвардса. В таких случаях в воспитание хоть и вносятся определенные изменения, зачастую удается добиться высокого качества жизни.
Во многих случаях лечение заболевания предполагает использование хирургических методик. Проводится оперативная коррекция пороков сердца, восстанавливается нормальная работа желудочно-кишечного тракта, ушиваются грыжи. Осуществляется и устранение косметических дефектов, таких как «заячья губа». На этапе лечения детям зачастую требуется зондовое питание, а также внутривенная инфузионная терапия. Поскольку при проведении хирургических вмешательств при синдроме Эдвардса велик риск развития инфекционных заболеваний, оправдано использование антибактериальных средств.
После радикальной коррекции врожденных нарушений потребуется длительная медикаментозная поддержка. Выбор тактики дальнейшего лечения основан на клинических проявлениях заболевания. Используются адреноблокаторы, мочегонные и гормональные препараты. Важную роль в реабилитации играет и правильное питание, которое зачастую возможно только при использовании смесей.
Профилактика
Специфических методов предотвращения развития заболевания не существует. Это связано с генетической природой поражения, а также неопределенностью его этиологии. Врачи рекомендуют женщинам старше 35 лет правильно планировать беременность, проводить регулярный скрининг. Наследственность редко играет роль в формировании проблемы, поскольку большинство пациентов с патологией не доживают до взрослого возраста. Исключение составляют лишь скрытые носители данной мутации. С целью определения особенности проводится анализ ДНК будущих родителей. Профилактика предполагает снижение вероятности возникновения нарушений в ходе митоза и мейоза клеток, что достигается за счет соблюдения принципов здорового образа жизни, а также поддержания должного уровня защитных сил организма.
Отзывы
Дарья, 37 лет, г. Псков
Дочь родилась с синдромом Эдвардса. У нее была неестественной формы головка, специфические черты лица. Врачи обнаружили также врожденный порок сердца. Ребенка забрали в отделение интенсивной терапии, где она пробыла три недели. Доктора сразу предупредили, что перспективы неутешительные. Дочка скончалась в возрасте месяца.
Кристина, 31 год, г. Казань
После наступления беременности регулярно ходила на осмотр к гинекологу. Во время очередного скринингового обследования на 12-й неделе врач в ходе УЗИ выявил отклонения, указывающие на синдром Эдвардса. Предупредил о рисках того, что ребенок родится больным. Мы с мужем решили оставить малыша. Однако на 20-й неделе произошел самопроизвольный аборт. Сейчас планируем новую беременность.
Загрузка...Синдром Эдвардса, ДНК-диагностика синдрома Эдвардса
Синдром Эдвардса (трисомия по хромосоме 18) - второе по частоте после синдрома Дауна хромосомное нарушение. Частота синдрома Эдвардса составляет 1:5000-1:7000 новорождённых. Девочки с синдромом Эдвардса рождаются в три раза чаще мальчиков.
| Рисунок 1. Пример диагностики синдрома Эдвардса методом КФ-ПЦР. | Рисунок 2. Больной ребенок с синдромом Эдвардса. |
 Пример диагностики синдрома Эдвардса (трисомии по хромосоме 18). Фиолетовым цветом выделены маркеры, расположенные в районе хромосомы 18, критическом для развития синдрома Эдвардса. В генотипе исследуемого образца по маркеру D18S978 наблюдается 1 пик (маркер неинформативен), по маркерам D18S535 и D18S386 - 3 пика (трисомия), по маркерам D18S390 и D18S819 - эффект дозы – неравное соотношение высоты двух пиков (трисомия). Таким образом, по четырем маркерам (D18S535, D18S386, D18S390 и D18S819) обнаружена трисомия, что позволяет установить диагноз «синдром Эдвардса». Генетический пол соответствует мужскому - присутствуют пики, соответствующие Y-хромосоме, по маркерам Amelogenin, 4SH, ZFXY, TAFL и присутствует пик гена SRY. Пример диагностики синдрома Эдвардса (трисомии по хромосоме 18). Фиолетовым цветом выделены маркеры, расположенные в районе хромосомы 18, критическом для развития синдрома Эдвардса. В генотипе исследуемого образца по маркеру D18S978 наблюдается 1 пик (маркер неинформативен), по маркерам D18S535 и D18S386 - 3 пика (трисомия), по маркерам D18S390 и D18S819 - эффект дозы – неравное соотношение высоты двух пиков (трисомия). Таким образом, по четырем маркерам (D18S535, D18S386, D18S390 и D18S819) обнаружена трисомия, что позволяет установить диагноз «синдром Эдвардса». Генетический пол соответствует мужскому - присутствуют пики, соответствующие Y-хромосоме, по маркерам Amelogenin, 4SH, ZFXY, TAFL и присутствует пик гена SRY. |
|
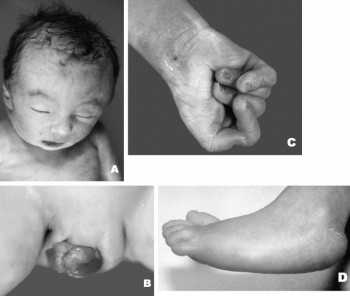
"Золотым стандартом" выявления хромосомных нарушений во всем мире долгое время являлся и продолжает оставаться метод кариотипирования с дифференциальной окраской хромосом. Этот метод позволяет анализировать кариотип в целом и определять крупные (не менее 5-10 млн пар нуклеотидов) хромосомные перестройки. Однако у него существует ряд ограничений, таких как трудоемкость, длительность (1-2 недели), высокие требования к квалификации и опыту специалиста, проводящего исследование, а также, в ряде случаев, технические проблемы (недостаточное количество и качество исследуемого материала, отсутствие митозов или роста культуры).
Этих недостатков лишен метод количественной флуоресцентной полимеразной цепной реакции (КФ-ПЦР), который все более широко применяется для диагностики анеуплоидий, в том числе и синдрома Эдвардса (Рис. 1). Этот метод обладает достоверностью, сравнимой с достоверностью стандартного кариотипирования, является более быстрым, дешевым, менее требовательным к количеству и качеству материала (поскольку не связан с ростом культуры клеток) и позволяет одновременно анализировать большое число образцов. Однако метод КФ-ПЦР имеет и ограничения: в мозаичных случаях он позволяет выявлять только высокоуровневый мозаицизм (от 20%), кроме того, он не может исключить наличие более редких хромосомных нарушений, которые могут быть связаны с пороками развития плода. При проведении дородовой диагностики синдрома Эдвардса, кроме материала плода, необходимо предоставлять биологический материал матери для того, чтобы исключить возможность получения ложноотрицательного результата из-за неправильного забора плодного материала. Анализ плодного материала выполняется за три рабочих дня.
При синдроме Эдвардса отмечается выраженная задержка пренатального развития, дети рождаются с пренатальной гипотрофией (средняя масса тела при рождении составляет 2340 г). Внешние проявления синдрома Эдвардса многообразны (Рис. 2). Наиболее типичными являются задержка психомоторного развития, гипоплазия скелетной мускулатуры и подкожной жировой ткани, врожденные пороки сердца, аномалии строения лица и черепа (долихоцефалия, микрофтальмия, укорочение глазных щелей, низкое расположение ушных раковин, микрогнатия, скошенный подбородок), множественные деформации кистей и стоп, аномалии развития желудочно-кишечного тракта, мочеполовой системы и центральной нервной системы (спинно-мозговые грыжи, гипоплазия мозолистого тела и мозжечка). Продолжительность жизни детей резко снижена: 90% из них погибают до года от осложнений, обусловленных врождёнными пороками развития (асфиксия, пневмония, кишечная непроходимость, сердечно-сосудистая недостаточность).
Причиной развития синдрома Эдвардса является утроение хромосомы 18. Трисомия по хромосоме 18 является частным случаем анеуплоидии– наличия в геноме набора хромосом, отличного от стандартного для данного вида и некратного ему. Трисомия хромосомы 18 обычно вызвана нерасхождением хромосом при формировании половых клеток родителя (яйцеклеток и сперматозоидов), в результате чего ребенок получает от матери или от отца лишнюю 18-ю хромосому. В этом случае все клетки организма ребёнка будут нести аномалию. В том случае, когда нерасхождение хромосом возникает при делении какой-либо клетки зародыша, наблюдается мозаичный вариант синдрома Эдвардса (10% случаев).
Риск рождения детей с синдромом Эдвардса, по разным литературным данным, не изменяется или незначительно возрастает с увеличением возраста беременной женщины.
Пренатальная диагностика синдрома Эдвардса включает в себя два этапа. На первом этапе, на сроке беременности 11-13 недель, проводится скрининг, который основывается преимущественно на биохимических показателях, поскольку на ранних сроках УЗИ не позволяет обнаружить в случае синдрома Эдвардса каких-либо грубых аномалий развития, которые могут быть выявлены лишь к 20-24 неделе. Биохимический анализ уровня определенных белков в крови беременной женщины (свободной β-субъединицы хорионического гормона человека (β-ХГЧ) и ассоциированного с беременностью плазменного протеина А (pregnancy associated plasma protein-A, РАРР-А)), с учетом ее возраста, позволяет рассчитать для нее риск рождения больного ребенка. Однако эти методы не позволяют поставить точный диагноз, и в результате проведенного скрининга лишь формируется группа риска беременных с повышенной вероятностью рождения больного синдромом Эдвардса. На втором этапе в группе риска проводится инвазивная процедура для получения плодного материала, необходимого для точного определения статуса плода. В зависимости от срока беременности это может быть биопсия ворсин хориона (8-12 недели), амниоцентез (14-18 недели) или кордоцентез (после 20-й недели). В полученных образцах ткани плода проводится определение хромосомного набора.
В Центре Молекулярной Генетики проводится диагностика синдрома Эдвардса (в том числе и пренатальная) методом КФ-ПЦР.
Эдвардса синдром